
Филогенетический анализ вирусов для
- 4059
- 3,4
- 0
- 5
Кладограмма бактерий и архей, построенная на основании 24-х генов с использованием Байесова вывода

Спонсор конкурса — дальновидная компания Thermo Fisher Scientific. Спонсор приза зрительских симпатий — фирма Helicon.
Что такое филогения и филогенетический анализ?
Филогения всех живых существ, или древо жизни, является нашим представлением о степени родства организмов и о том, как шла эволюция живых существ. Кто является ближайшим родственником человека, и каким был наш общий предок? Вымерли ли динозавры, или их потомки до сих пор живут рядом с нами? Произошли ли теплокровность и способность к полету среди позвоночных единожды? Откуда вообще взялись позвоночные? На все эти вопросы уже есть ответы, и получены они были главным образом с помощью филогенетического анализа.
Немного о ДНК
Чтобы понять, как анализируют ДНК, надо вспомнить, как она устроена. ДНК, или дезоксирибонуклиновая кислота, — это очень длинная молекула, которая находится в ядре клетки. ДНК, как правило, состоит из двух закрученных спиралей, а каждая спираль состоит из множества нукеотидов. Нуклеотиды по большей части отличаются друг от друга азотистыми основаниями, которых в ДНК всего четыре: аденин, тимин, гуанин и цитозин. Именно нуклеотиды создают слабые химические связи, которыми соединяются спирали ДНК. Аденин одной спирали связывается с тимином другой спирали, а гуанин связывается с цитозином (рис. 1). Мутация происходит, когда одно основание заменяется на любое другое. Чаще всего замены происходят в парах аденин—гуанин и тимин—цитозин.
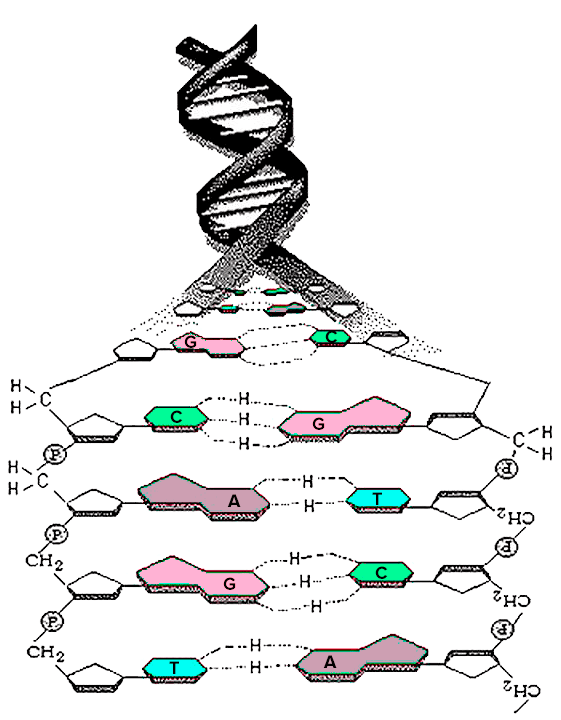
Рисунок 1. ДНК. A — аденин, C — цитозин, G — гуанин, T — тимин.
В ДНК есть последовательности нуклеотидов, которые кодируют белки, и есть участки, которые ничего не кодируют. Кодирующие последовательности — это гены. Они могут быть разной длины, но чаще всего имеют определенную структуру, по которой можно сказать — ген это или нет. Именно гены обычно используют для филогенетического анализа.
Основные принципы построения филогений
Методы построения филогений еще в 60-х годах XX века разделились на две основные ветви — фенетические и кладистические. В то время анализ родственных связей основывался на морфологических признаках [12]; с привлечением к построению филогений молекулярных признаков основные принципы анализа родственных связей остались фактически теми же.
- В фенетике построение филогении основано на общем сходстве двух видов — то есть, чем больше общих признаков, тем ближе они друг к другу;
- В кладистике же считается, что только уникальные для какой-либо группы признаки можно использовать для оценки родства таксонов. Родоначальником кладистического анализа является немецкий ученый Вилли Хенниг [6]. Этот автор также ввел и терминологию, которая широко используется до сих пор. Уникальные признаки называются апоморфиями; ветви, которые объединяются апоморфиями — это клады; а сама филогения называется кладограммой (рис. 2) [12].
Чтобы было более понятно, представьте три вида животных: домашнюю мышь, сумчатую мышь и кенгуру. Домашняя мышь и сумчатая мышь очень похожи друг на друга внешне, но у сумчатой мыши и кенгуру есть общая апоморфия — сумка, — что говорит о том, что эти два вида родственные. Но, естественно, филогенетический анализ основывается на гораздо большем количестве признаков, и группы могут иметь несколько апоморфий.
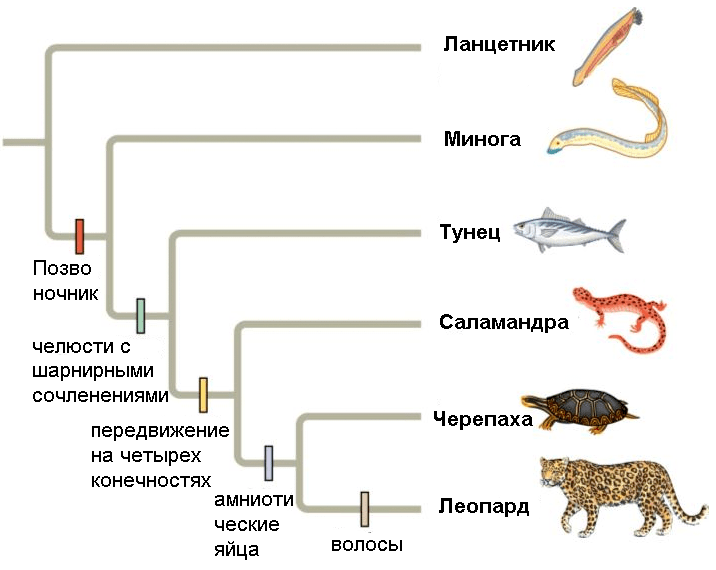
Рисунок 2. Полностью разрешенная кладограмма. Каждое ветвление — это клада. Обозначенные признаки являются апоморфиями.
Первые шаги. ДНК—ДНК гибридизация
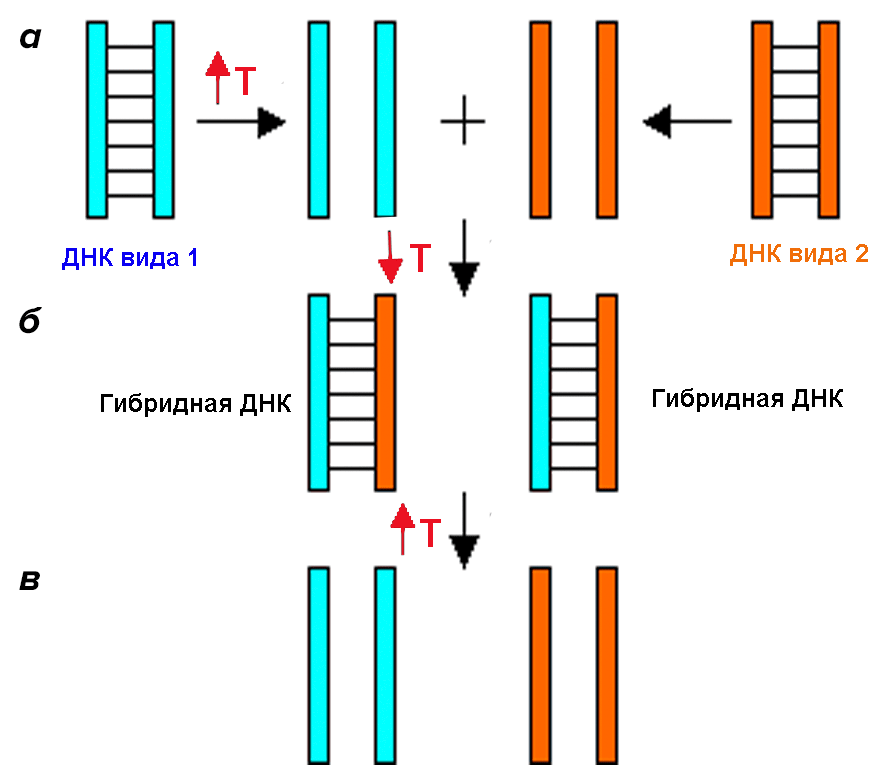
Рисунок 3. ДНК—ДНК гибридизация. а — Нагревание ДНК двух видов, в результате которого двойная спираль распадается на две части. б — Охлаждение ДНК, в результате которого молекулы ДНК разных видов гибридизуются друг с другом. в — Нагревание ДНК, в результате которого гибридные молекулы ДНК распадаются.
ThinkQuest, рисунок с изменениями
Очень быстро стало понятно, что такой метод не может быть очень точным. Дело в том, что гены могут гибридизоваться не только с гомологичными им генами (гены-ортологи), но и с копиями этих генов, которых в геноме может быть довольно много (гены-паралоги) [15]. Постепенно, с развитием методики секвенирования генов , главным источником для построения филогений стали последовательности ДНК или белков, записанные в виде компьютерных файлов. В последние годы скорость накопления генетической информации растет все увеличивающимися темпами, что окончательно утверждает филогению как метод анализа и обработки биологических текстов.
Метод матрицы расстояний (distance matrix)
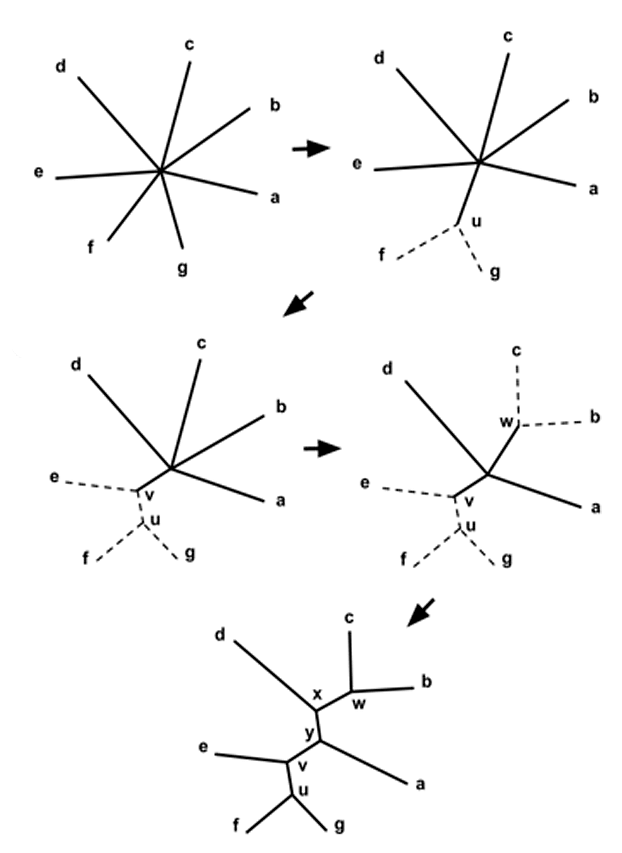
Рисунок 4. Метод ближайшего соседа
Разные авторы, однако, перечисляют некоторые минусы метода ближайших соседей. Например, есть мнение, что этот метод хуже работает с таксонами, которые филогенетически далеки друг от друга [4], [17]. Также недостатком можно считать и то, что метод всегда выдает дерево с одним-единственным возможным вариантом ветвления [3]. Это происходит потому, что алгоритм подразумевает построение одной филогении без сравнения с другими, тогда как в кладистических методах оцениваются деревья с различным порядком ветвления. Несмотря на то, что в серьезных филогенетических анализах методы матрицы расстояний сейчас почти не используются, они применяются, например, для быстрого построения филогений близкородственных бактерий и вирусов [18].
Метод наибольшей экономии (maximum parsimony)
Эволюция признака — тоже событие нечастое, и когда мы видим два похожих по строению органа, то мы предполагаем, что орган произошел один раз [3]. Это не означает, что признак действительно произошел только один раз, просто это наиболее вероятно. Кладограмма строится на основании многих признаков, и чем больше апоморфий характеризует ту или иную ветвь, тем больше доверия она вызывает.
Другой минус в том, что метод не учитывает разные модели замены нуклеотидов [17]. Например, в методе наибольшей экономии аденин имеет одинаковую вероятность уступить место как тимину, так и цитозину, хотя, как уже отмечалось выше, в организме аденин скорее заменится на цитозин, чем на тимин.
Методы, основанные на моделях эволюции
Наиболее часто используемые методы построения филогений на основе молекулярных данных основываются на моделях эволюции. Один из первых стал метод максимального правдоподобия (maximum likelihood). Для расчета кладограммы, помимо последовательности ДНК, надо выбрать модель замены нуклеотидов, на основании которой будут рассчитываться вероятности. Также в расчет берется длина ветви или эволюционная дистанция между двумя таксонами. Во время анализа рассчитывается, какая длина ветви наиболее вероятна с точки зрения выбранной модели, вероятности всех ветвей кладограммы умножаются, и кладограмма, имеющая наибольшую вероятность, считается правильной [3], [16], [17].
Последний и, наверное, самый популярный в наше время метод — это Байесовский вывод (Bayesian inference). Он, в общем, похож на метод максимального правдоподобия, поскольку также основывается на модели и длине ветвей. Но отличие Байесовского вывода в том, что тут берется в расчет еще один фактор — апостериорная вероятность (posterior probablity), которая рассчитывается на основании как исходных данных, так и полученных результатов анализа [3], [16], [17]. Это не очень понятно интуитивно, но суть в том, что в ходе анализа исследователь получает новые данные, которые тоже можно применить.
Приведу очень простой пример. Пусть у нас есть мешок с сотней шариков, половина их которых красные и половина — белые. Изначально вероятность вытащить шарик как белого, так и красного цвета равна 50%. Но, допустим, мы вытащили 20 красных и 40 белых шариков, и в мешке остались 30 красных и 10 белых шариков. Это означает, что к текущему моменту шанс вытащить красный шарик равен 75%, а белый — 25%, что кардинальным образом отличается от исходного состояния. В Байесовском выводе используются похожая логика, хотя, конечно же, расчеты там гораздо сложнее.
Все же насколько достоверны филогении?
Думаю, что внимательный читатель заметил, что многие перечисленные методы основаны на вероятностях, и у него может возникнуть закономерный вопрос: как можно доверять филогении, если всегда есть шанс, что построенное дерево ошибочно и не соответствует действительному ходу эволюции? Действительно, методы несовершенны, но на этот вопрос ответ есть.
Во-первых, в филогенетических методах есть понятие «поддержка: чем больше уникальных признаков поддерживают дерево или какую-то его ветвь, тем больше доверия они вызывают [12]. Само дерево может иметь низкую поддержку, зато свидетельств в пользу отдельных его ветвей может быть так много, что корректность не вызовет сомнений. Для подтверждения результата исследователи могут использовать совокупности признаков: последовательности ДНК, РНК и белков, морфологические данные, особенности поведения организмов и многое другое [11]. Когда независимые признаки подтверждают друг друга, уверенность в результате гораздо выше.
Второй ответ на поставленный вопрос еще более обнадеживающий. Его дают эксперименты, проведенные на разных организмах, для которых известна генеалогия, то есть настоящая эволюционная история [1], [5], [7], [10]. Можно привести в пример опыт с мышами, когда филогенетический анализ провели на основе ДНК 24-х линий этих животных. Оказалось, что наблюдаемая последовательность поколений и полученная филогения почти полностью соответствуют друг другу [1]. Это значит, что используемые методы как минимум способны правильно отображать эволюцию.
Плюсы и минусы молекулярных методов построения филогений
У молекулярных методов есть много преимуществ перед морфологическим анализом. Во-первых, ДНК содержит в себе множество данных, которые можно использовать в расчетах, — ведь в генах могут содержаться сотни нуклеотидов. Чаще всего для оценки родства используют больше одного гена, тогда как для анализа на основе морфологических данных используют несколько десятков признаков. Во-вторых, анализ ДНК считается более объективным. Дело в том, что морфологические признаки разные люди могут трактовать и кодировать по-разному, тогда как нуклеотиды всегда одинаковы . В-третьих, ДНК можно использовать как для анализа групп высоких рангов, так и для выяснения отношений между видами, и даже между отдельными индивидами. Морфологический же анализ более достоверен при работе с таксонами высоких рангов, чем на уровне видов, — просто потому, что чем выше ранг, тем лучше отличаются группы, и тем легче отличить аналогичный признак от гомологичного.
Первая причина заключается в том, что не каждый организм подходит для выделения ДНК. Он должен быть собран и сохранен специальным образом, иначе эта молекула просто разрушается. Множество редких и интересных видов было описано много десятков лет назад, когда еще даже про ДНК ничего не знали, и в наши дни не очень понятно, где их искать и как собирать. В первую очередь это касается мелких членистоногих, — особенно насекомых, которых чаще всего хранят сухими. То же самое можно сказать и о палеонтологических находках вымерших видов. Для оценки родства таких групп можно использовать только морфологические методы.
Третья причина — это высокая стоимость секвенирования генов. Для построения филогении одного небольшого рода можно легко потратить пару тысяч долларов. А если учесть, что гены не всегда подбирают правильно с первого раза, или некоторые экземпляры оказываются непригодными для секвенирования, то анализ надо проводить повторно, и цена может быть больше, чем предполагалось изначально. Анализ же на основе морфологических признаков обходится гораздо дешевле.
Вирус болезни Ньюкасла (БН) широко распространенное заболевание, регистрируемое во многих странах и поражающее многие виды диких и домашних птиц.
Несмотря на значительные успехи в диагностике, эпизоотологии, изучении патогенности вируса БН на молекулярном уровне, проблема борьбы с этим особо опасным заболеванием птиц остается актуальной [1, 2]. В основе профилактики болезни Ньюкасла лежат неспецифические и специфические средства защиты и методы их осуществления [3, 4].
Возбудитель БН – РНК-содержащий вирус (парамиксовирус птиц типа 1) являющийся таксоном рода Avulavirus, подсемейства Paramyxovirinae, семейства Paramyxoviridae, порядка Mononegavirales и характеризующийся минус-нитевым РНК-геномом. Его вирионная РНК представлена шестью генами, кодирующими гемагглютинин-нейраминидазу (HN), нуклеопротеин (NP), фосфопротеин (P), матриксный белок (М), РНК-зависимую РНК-полимеразу (L) и белок слияния (F), и двумя неструктурными белками V и W [5].
Для идентификации изолятов вируса болезни Ньюкасла были использованы полимеразная цепная реакция в реальном времени (РВПЦР) и антигенный анализ с помощью моноклональных антител. Реагенты для реакции были предоставлены Шведским ветеринарным университетом (SVA) города Упсала.
Для выделения вируса использовали клоакальные смывы и кусочки органов от домашних и диких птиц. До начала исследований пробы хранили в низкотемпературном морозильнике (±70 ºС).
РНК вируса БН выделяли с помощью коммерческого набора RNeasy Mini Kit (QIAGEN, Германия) согласно инструкции. Реакционный объем составил 20 мкл, а концентрация праймеров 10 мкмоль на реакцию. Постановка реакции была проведена согласно протоколу к диагностическому набору. Был проведен 1 цикл в режиме 50 °С – 15 мин, 95 °С – 5 мин и 45 циклов амплификации в режиме 90 °С – 30 сек, 60 °С – 60 сек. РВПЦР проводили в приборе Rotor Gene 6 000 (Software). Результаты проведенной РВ-ПЦР приведены в таблице.
Таблица. Результаты исследованных образцов БН в РВ-ПЦР
| Область | Вид птиц | Количество исследованны х птиц | Трахеальный смыв | Клоакальный смыв | ||
| количество образцов | результат | количество образцов | результат | |||
| Джамоат Мирзо Ризо, г. Гиссар | утка | 2 | 2 | отрицательный | 2 | отрицательны й |
| цыпленок | 2 | 2 | положительный | 2 | отрицательны й | |
| голубь | 2 | 2 | отрицательный | 2 | отрицательны й | |
| куропатка | 1 | 1 | положительный | 1 | отрицательны й | |
| индюк | 1 | 1 | положительный | 1 | отрицательны й | |
| Джамоат Чимтепп а | дикий голубь | 3 | 3 | положительный | 3 | отрицательны й |
Как видно из таблицы, из 22 исследованных проб в 2-х выявлены РНК вируса болезни Ньюкасла. При этом положительный результат отмечен только у кур. Все пробы уток, голубей, куропаток, индюков из Гиссарского района и диких голубей из джамоата Чимтеппа района Рудаки дали отрицательный результат в РВ-ПЦР. Результаты ПЦР в реальном времени приведены на рис. 1.
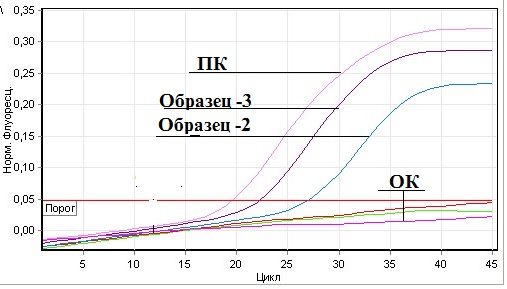
Рис.1. Результаты РВ-ПЦР на вирус БН
Премечание: ПК – положительный контроль; ОК — отрицательный контроль; Образец-2 – трахеальный смыв цепленка 1.1; Образец-3 – трахеальный смыв цепленка 2.1.
Совместно со специалистами Шведского национального ветеринарного университета (SVA, Упсала) изучено филогенетическое дерево изолята вируса болезни Ньюкасла, выделенного в Таджикистане.
При скрининге материалов Гиссарского района в ПЦР получены положительные результаты с обнаружением специфических продуктов 300 пар оснований, что свидетельствует о наличии РНК вируса БН в исследованных образцах.
После выделения вируса проведено секвенирование фрагмента гена, кодирующего белок слияния (F-протеин). В результате секвенирования расшифрована последовательность нуклеотидов, которая включает сайт расщепления белка слияния, что важно для выявления его патогенности.
Для определения филогенетических взаимоотношений секвенированного вируса осуществлен анализ имеющихся в международной базе данных полных нуклеотидных последовательностей F-гена (GGRRQKRFIGAV) ПМВ-1.
Результаты молекулярно-биологического исследования в полимеразной цепной реакции с обратной транскрипцией (ОТ-ПЦР) и методом нуклеинового секвенирования генома позволили идентифицировать изолят от домашних птиц, относящийся к авулавирусу птиц 1- серотипа,7i — генотипа (VIIi), т.е. вирус БН (Avian Avulavirus type 1) (рис. 2).
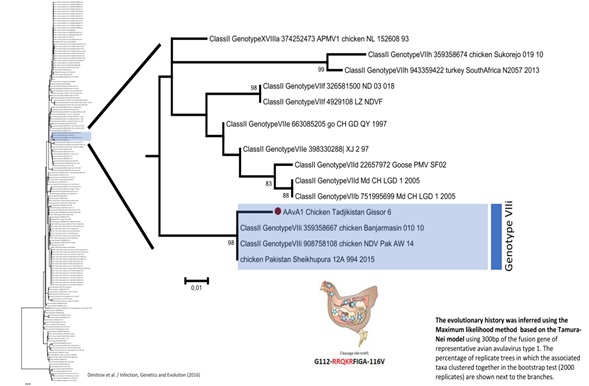
Рис. 2. Дендрограмма Гиссарского изолята вируса болезни Ньюкасла по антигенному родству выполненная в Шведском национальном ветеринарном университете (SVA), Упсала.
Как видно из рис. 2, Гиссарский изолят вируса болезни Ньюкасла (на рисунке текст выделен) по антигенному варианту совпадает на 98 % с штаммами стран южной Азии, такими как Шри Ланка и Пакистан.
Заключение
Таким образом, с помощью ОТ-ПЦР и моноклинальных антител показана степень антигенного родства вируса болезни Ньюкасла, циркулирующего в Таджикистане, с вирусами из других стран.
Авторы
Д. М. Шоназар, кандидат ветеринарных наук, заведующий лабраторией диагностики вирусных болезней животных и птиц Института проблем биологической безопасности ТАСХН (ИПББ), Душанбе
Г. Н. Мамадатохонова, старший научный сотрудник Института проблем биологической безопасности ТАСХН (ИПББ), Душанбе
С. Зохари, микробиолог, Шведский национальный ветеринарный университет (SVA), Упсала
Библиографические ссылки
1. Безбородова Н. А., Ким Н. А.Сравнение лабораторных методов диагностики инфекций, вызываемых патогенными и условно-патогенными микроорганизмами // Эффективное животноводство. 2018. № 2 (141). С. 46–47.
2. Методы клинико-лабораторной диагностики острых респираторных вирусных инфекций у крупного рогатого скота / Порываева А. П., Печура Е. В., Вялых И. В., Томских О. Г., Бусыгина Н. С. // Вопросы нормативно-правового регулирования в ветеринарии. 2017. № 3. С. 55–58.
3. Alexander D. J. Newcastle disease and other avian paramyxoviruses Rev Sci Tech, 2000. № 19 (2). Р. 443–462.
4. Kaverin N. V., L’vov D. K. Paramiksovirusy (Paramyxoviridae) [Paramyxoviruses]. V kn.: Medicinskaja virusologija, 2008. Р. 183–189.
Аннотация научной статьи по биологическим наукам, автор научной работы — И.С. Клименко, Н.В. Хуторецкая, Т.В. Гребенникова
Проведено молекулярно-генетическое исследование штаммов вирусов москитных лихорадок, выделенных на территории Афганистана в 1986—1987 гг. С помощью универсальных праймеров была проведена ОТ-ПЦР и секвенирование фрагментов S сегмента генома изучаемых вирусов. Полученные нуклеотидные последовательности длиной 379 н.о. были использованы для филогенетического анализа афганских штаммов. Установлено, что афганский штамм Аф-1008 является представителем неаполитанского серокомплекса, а штамм Аф-1028 — сицилийского.
Похожие темы научных работ по биологическим наукам , автор научной работы — И.С. Клименко, Н.В. Хуторецкая, Т.В. Гребенникова
PHYLOGENETIC ANALYSES OF THE STRAINS OF SANDFLY FEVER VIRUSES ISOLATED IN AFGHANISTAN
Molecular-biological examination of the strains of sandfly fever viruses isolated in Afghanistan in 1986—1987 years was carried out. It was make by RT-PCR and sequenation of fragment of S-segment of genome studied viruses with universal primers. Received nucleotide sequences of the length 379 p.n. have been used for phylogenetic analyses Afghanistan strains. It was established that Afghanistan strain Af-1008 is member of Naples serogroup and strain Af-1028 is member of Sicilian serogroup.
ФИЛОГЕНЕТИЧЕСКИЙ АНАЛИЗ ШТАММОВ ВИРУСОВ МОСКИТНЫХ ЛИХОРАДОК, ВЫДЕЛЕННЫХ НА ТЕРРИТОРИИ АФГАНИСТАНА
И.С. Клименко, Н.В. Хуторецкая
ГУ НИИ вирусологии им. Д.И. Ивановского РАМН ул. Гамалеи, 16, Москва, Россия, 123098
Медицинский факультет Российский университет дружбы народов ул. Миклухо-Маклая, 6, Москва, Россия, 117198
Проведено молекулярно-генетическое исследование штаммов вирусов москитных лихорадок, выделенных на территории Афганистана в 1986—1987 гг. С помощью универсальных праймеров была проведена ОТ-ПЦР и секвенирование фрагментов S сегмента генома изучаемых вирусов. Полученные нуклеотидные последовательности длиной 379 н.о. были использованы для филогенетического анализа афганских штаммов. Установлено, что афганский штамм Аф-1008 является представителем неаполитанского серокомплекса, а штамм Аф-1028 — сицилийского.
Вирусы москитных лихорадок (МЛ), относящиеся к роду Phlebovirus (Bunya-viridae), были впервые выделены А.В. Sabin из крови больных в период эпидемии среди американских солдат в 1943—1944 гг. в Италии: вирус сицилийской москитной лихорадки (СМЛ) — в Палермо (Сицилия), вирус неаполитанской москитной лихорадки (НМЛ) — в Неаполе [6]. Вирусы МЛ вызывают заболевания человека с клиническими синдромами, варьирующими от классической лихорадки до менингоэнцефалита [1, 2]. Вирусы в Старом свете циркулируют в пределах обитания москитов, среди которых основными переносчиками являются Phlebo-tomus papatasii. Это территория Восточного полушария между 20 и 40 градусами с.ш.: Азия (Иран, Узбекистан, Таджикистан, Туркменистан), Восточная Европа (Югославия, Молдавия, Израиль, юг европейской части России и Украины), средиземноморские страны (Африка, Южная Европа) [1, 7, 6].
В мае—августе 1986—1987 гг. во время военных действий в Афганистане от советских военнослужащих с признаками лихорадки были выделены новые штаммы вирусов москитных лихорадок, представленные, согласно серологическим данным, вариантом неаполитанского серокомплекса (Аф-1008) и сицилийского серокомплекса (Аф-1028) [10].
Геном флебовирусов состоит из трех кольцевых одноцепочечных сегментов РНК отрицательной полярности (большой L, средний М и малый S), каждый из которых ответственен за синтез определенных вирусных белков. В течение последних лет определены полноразмерные нуклеотидные последовательности или участки сегментов генома флебовирусов, на основе которых проводится филогенетический анализ [3].
Цель настоящего исследования — проведение филогенетического анализа штаммов вирусов МЛ из коллекции лаборатории биологии и индикации арбови-русов ГУ НИИ вирусологии им. Д.И. Ивановского РАМН, выделенных на территории Афганистана, на основе нуклеотидных последовательностей фрагментов S-сегмента генома, полученных методом ОТ-ПЦР.
Материалы и методы. В работе использовались штаммы вирусов москитных лихорадок из коллекции лаборатории биологии и индикации арбовирусов ГУ НИИ вирусологии им. Д.И. Ивановского (табл. 1). Все вирусы пассировались на двухдневных мышах-сосунках.
Штаммы вирусов москитных лихорадок, используемые в работе
Серокомплекс Штамм Место выделения Источник выделения Год выделения
Неаполитанская москитная лихорадка Sabin Неаполь, Италия человек 1944
1008 Афганистан человек 1986—1987
Сицилийская москитная лихорадка Sabin Сицилия, Италия человек 1943
1028 Афганистан человек 1986—1987
Тоскана ISS Phl 3 Тоскана, Италия Phlebotomus perniciosus 1971
Каримабад !-58 R-12928 Каримабад, Иран Phlebotomus papatasi 1972
Конструирование праймеров. Нуклеотидные и аминокислотные последовательности S-сегмента генома вирусов МЛ, представленные в базе данных NCBI GenBank, были выровнены с помощью программы CLUSTAL W. Последовательности специфических олигонуклеотидов разрабатывали в программе DNASTAR V.3.12 (Lasergene, США) на основе усредненной последовательности S-сегмента вирусов МЛ (табл. 2).
Праймеры, использованные для проведения ОТ-ПЦР фрагментов S-сегмента генома вирусов москитных лихорадок
Праймер Назначение Длина (н.о.) Нуклеотидная последовательность 5'—3'
Unique FS ПЦР, секвенирование 20 GCGAAGCTAGGGTGCATCAT
Unique R_S ПЦР, секвенирование 20 TTTGCTTACCAGGGGTTTGA
Анализ фрагментов ДНК после ПЦР проводили методом электрофореза в ага-розном геле (2%). Гели фотографировали и анализировали, используя трансиллюминатор, в ультрафиолетовом свете с длиной волны 254 нм.
Праймеры для секвенирования были те же, что и для ОТ-ПЦР. Определение первичной нуклеотидной последовательности осуществляли на автоматическом секвенаторе ABI PRISM 3130 (Applied Biosystems, США) согласно рекомендациям производителя.
Результаты. Для конструирования праймеров последовательности S-сегмента флебовирусов (СМЛ, НМЛ, Тоскана и Каримабад), представленные в базе данных NCBI GenBank, были выровнены, как описано выше. На усредненной нук-леотидной и аминокислотной последовательностях были определены наиболее консервативные участки для расположения специфических олигонуклеотидов. Выбранные нами праймеры Unique F_S и Unique R_S амплифицировали все используемые в работе штаммы вирусов МЛ, указанных в табл. 1. Методом ОТ-ПЦР получили фрагменты S-сегмента генома изучаемых нами штаммов длиной 379 н.о. Определили их первичную нуклеотидную последовательность.
Далее был выполнен филогенетический анализ полученных последовательностей фрагмента S-сегмента генома исследуемых штаммов в сравнении с ранее опубликованными последовательностями флебовирусов.
На рис. 1 показана филогенетическая дендрограмма. Исследуемые штаммы четко разделились на две группы, обозначенные на рисунке цифрами I и II и соответствующие неаполитанскому и сицилийскому серокомплексу. Вирус Карима-бад, как видно из рис. 1, не был включен ни в одну из них. Афганский штамм Аф-1008 вошел в состав группы I, а Аф-1028 — группы II.
Обсуждение. L, M и S сегменты генома флебовирусов играют различную роль в вирусном патогенезе. РНК малого сегмента проявляет двойную кодирующую стратегию и содержит две открытые рамки считывания.
Одна из них кодирует нуклеокапсидный белок N, а другая — неструктурный белок NSs [5]. Известно, что N белок проявляет комплементсвязывающую активность [10]. Согласно большинству недавних исследований, S-сегмент флебовирусов наиболее консервативен по сравнению с М-сегментом генома [4, 8]. Имеющиеся в коллекции лаборатории биологии и индикации арбовирусов ГУ НИИ вирусологии им. Д.И.Ивановского афганские штаммы вирусов МЛ (1008 и 1028) были охарактеризованы лишь серологически. На основании результатов серологической идентификации в РСК в составе серокомплекса НМЛ были выделены три антигенные группы: 1 — вирусы НМЛ, Тоскана и Тегеран, 2 — афганские штаммы
Аф-130, Аф-1038, Аф-1008. Вирус Каримабад оказался по данным этого теста полностью изолированным от серокомплекса. Афганские штаммы Аф-83 и Аф-1028 по результатам исследований оказались идентичными друг другу и прототипному штамму СМЛ. В то же время они не вступали в перекрестные взаимодействия с представителями неаполитанского серокомплекса [9, 10]. Для того, чтобы дать генетическую характеристику штаммам Аф-1008 и Аф-1028, мы решили амплифи-цировать, а затем секвенировать участок S-сегмента генома этих вирусов. Мы попытались найти универсальную пару праймеров, которая амплифицировала один и тот же участок сегмента у нескольких вирусов. Как видно из рис. 1, распределение изучаемых вирусов четко соответствует их серологической группировке. Группу I составили вирусы неаполитанского серокомплекса, который разделился на две ветки: вирус нмл^ып) и НМЛ-подобные вирусы, вирус Тоскана и Тоскана-подобные вирусы. Группу II образовали вирус СМЛ и его различные штаммы. Вирус Каримабад оказался обособлен от вышеперечисленных групп.
Рис. 1. Филогенетическая дендрограмма вирусов москитных лихорадок на основе участка S-сегмента длиной 379 н.о.
Нуклеотидная последовательность участка S сегмента прототипного штамма НМЛ, секвенированного нами, оказалась идентичной (100%) таковой, опубликованной в базе данных GenBank. То же можно сказать и про прототипный штамм вируса Тоскана (100%). Афганский штамм Аф-1008 вошел в состав неаполитанского серокомплекса, а наиболее близким ему оказался штамм НМЛ индийского происхождения (Р-7101795) — 93%.
Исследуемый нами афганский штамм Аф-1028, согласно полученным данным, относится к сицилийскому серокомплексу. Причем наиболее близким ему филогенетически оказался индийский штамм СМЛ (I 701735) — 97%.
Таким образом, нами впервые секвенированы афганские штаммы вирусов неаполитанской и сицилийской москитных лихорадок и были установлены данные о систематическом положении изученных штаммов вирусов внутри антигенной группы.
[1] Bartelloni P.J., Tesh R.B. Clinical and serological responses of volunteers infected with phle-botomus fever virus (Sicilian type) // Am. J. Med. Hyg. — 1976. — N 25. — P. 456—462.
[2] Braito A., Ciufolini M.G., Pippi L., Corbisiero R., Fiorentini C., Gistri A., Toscano L. Phle-botomus-transmitted Toscana virus infection of the central nervous system: a seven-year experience in Tuscany // Scand. J. Infect. Dis. — 1998. — N 30. — P. 505—508.
[3] Giorgi C., AccardiL., Nicoleti L., Gro M.C., Takehara K., Hilditch C., Morikawa S., Bishop D.H. Sequences ahd coding strategies of the S RNAs of Toscana and Rift Valley fever viruses compared to those of Punta Toro, Sicilian Sandfly fever, and Uukuniemi viruses // Virology. — 1991. — N 180. — P. 738—753.
[4] Liu D.Y., Tesh R.B., Travassos Da Rosa A.P., Peters C.J., Yang Z., Guzman H., Xiao S.Y. Phy-logenetic relationships among members of the genus Phlebovirus (Bunyaviridae) based on partial M segment sequence analyses // J. Gen. Virol. — 2003. — N 84. — P. 465—473.
[5] Nichol S.T., Beaty B.J., Elliot R.M., Goldbach R., Plyusnin A., Schmaljohn C.S., Tesh R.B. Genus Phlebovirus. In Virus Taxonomy: Eighth Report of the International Committee on Taxonomy of Viruses. — 2005 — P. 709—711.
[6] Tesh R.B., Saidi S., Gajdamovich S.J., Rodhain F., Vesenjak-Hirjan J. Serological studies on the epidemiology of sandfly fever in the Old World // Bull WHO. — N 54. — P. 663—674.
[7] Vesenjak-Hirjan J., Punda-Polic V., Dobe M. Goegraphical distribution of arboviruses in Yugoslavia // J. Hyg. Epidemiol. Microbiol. Immunol. — 1991. — Vol. 35. — N 2. — P. 129—140.
[8] Xu F., Chen H., Travassos da Rosa A.P., Tesh R.B., Xiao S.Y. Phylogenetic relationships among sandfly fever group viruses (Phlebovirus: Bunyaviridae) based on the small genome segment // J. Gen. Virol. — 2007. — N 88. — P. 2312—2319.
[9] Белан Э.Б. Биологические и антигенные свойства штаммов вирусов сицилийской и неаполитанской москитных лихорадок, выделенных в Афганистане в 1986—1987 гг.: Диса . канд. мед. наук. — М., 1994.
[10] Гайдамович С.Я., Хуторецкая Н.В., Мельникова Е.Е. и др. Вирусологическое исследование случаев москитных лихорадок в Афганистане // Вопросы вирусологии. — 1990. — N 35(1). — C. 45—47.
PHYLOGENETIC ANALYSES OF THE STRAINS OF SANDFLY FEVER VIRUSES ISOLATED IN AFGHANISTAN
I.S. Klimenko, N.V. Khutoreckaya
D.I. Ivanovsky Institute of virology, RAMSci, Gamaleya str., 16, Moscow, Russia, 123098
Medical faculty Peoples' Friendship University of Russia
M-Maklaya str., 8, Moscow, Russia, 117198
Molecular-biological examination of the strains of sandfly fever viruses isolated in Afghanistan in 1986—1987 years was carried out. It was make by RT-PCR and sequenation of fragment of S-segment of genome studied viruses with universal primers. Received nucleotide sequences of the length 379 p.n. have been used for phylogenetic analyses Afghanistan strains. It was established that Afghanistan strain Af-1008 is member of Naples serogroup and strain Af-1028 is member of Sicilian serogroup.
ФАКТИЧЕСКОЕ ПИТАНИЕ КОРЕННОГО МАЛОЧИСЛЕННОГО НАСЕЛЕНИЯ РЕСПУБЛИКИ САХА (ЯКУТИЯ)
Т.М. Климова, М.Е. Балтахинова, В.И. Федорова, В.Г. Кривошапкин
Изучение среднесуточного потребления белков, жиров, углеводов, основных минеральных веществ и витаминов среди коренного малочисленного населения Республики Саха (Якутия). Эпидемиологическое исследование проведено среди населения 3 районов Республики Саха (Якутия), в местах компактного проживания коренных малочисленных народов. Исследование фактического питания проведено методом 24-часового воспроизведения. В исследовании приняли участие 696 человек в возрасте 20—79 лет (отклик 85%).
Результаты исследования и выводы. Выявлена несбалансированность рациона питания: избыточное потребление простых углеводов, глубокий дефицит минеральных веществ и витаминов.
Ключевые слова: питание, витамины, минеральные вещества.
Читайте также:
