
Системная красная волчанка и системная склеродермия что это такое
Последнее обновление: 15.01.2020
КРАСНАЯ ВОЛЧАНКА(lupus erythematodes; син. эритематоз) - заболевание из группы диффузных болезней соединительной ткани (коллагенозов). Различают две основные формы красной волчанки: кожную и системную. Кожная форма обычно проявляется в виде дискоидной красной волчанки, диссеменированной и глубокой красной волчанки.
Заболевание является мультифакториальным. Имеет значение наследственная предрасположенность. В патогенезе определенную роль играют инсоляция, хроническая очаговая (чаще стрептококковая) инфекция, переохлаждение, гормональный дисбаланс, лекарственная непереносимость, аутоаллергия.
Клиника. Для кожного поражения характерны три кардинальных симптома:
- инфильтративная эритема,
- фолликулярный гиперкератоз,
- рубцовая атрофия.
Дискоидная красная волчанка наиболее часто встречается в странах с влажным и холодным климатом. Среди больных преобладают женщины молодого и среднего возраста.

Типична локализация сыпи на коже лице (особенно на носу и щеках, где очаг может напоминать по форме бабочку), ушных раковинах. Нередко поражается волосистая часть головы и красная кайма губ, причём при поражении губ процесс выходит за пределы красной каймы. Возможно поражение слизистой оболочки рта.
При переходе в атрофическую стадию в центре очага формируется рубцовая атрофия, в зоне которой могут быть телеангиоэктазии и краевая пигментация, а на коже волосистой части головы - рубцовое облысение. Очаги поражения медленно увеличиваются.
Характерно длительное непрерывное течение с периодическими ухудшениями в весенне-летний период. Признаки системности процесса обнаруживаются крайне редко. Прогноз для жизни благоприятный, но заболевание приводит к грубым косметическим дефектам.
При диссеминированной красной вволчанке характерно образование нескольких очагов поражения кожи. Эритема, инфильтрация, фолликулярный гиперкератоз выражены гораздо слабее, чем при дискоидной красной волчанке. Помимо кожи головы очаги диссеменированной красной волчанки выявляются на верхних частях груди и спины, на тыле кистей. Поражение обычно не приводит к формированию грубых косметических дефектов, однако обследование в динамике нередко позволяет выявить признаки системности.
Промежуточное место между кожными и системнымими формами красной волчанки занимает так называемая подострая кожная форма, для которой характерны распространённые кольцевидные очаги на коже, которые при слиянии образуют полициклические, шелушащиеся по краям участки на груди, спине и конечностях с гиперпигментацией телеангиоэктазиями в центре. При этой форме заболевания выявляют умеренно выраженные признаки системного процесса: артралгии, анемия, лейкоцитопения, тромбоцитопения, может выявляться антинуклеарный фактор, антитела к ДНК, однако в отличие от системной красной волчанки прогноз заболевания относительно благоприятный.
При глубокой красной волчанке наряду с описанными ранее изменениями кожи в подкожной клетчатке имеется один или несколько резко отграниченных плотных, подвижных узловатых уплотнений, так называемый люпус-панникулит.
Лечение кожной формы красной волчанки должно быть комплексным.
- Применяют глюкокортикоиды, цитостатики.
- Рекомендуется санация выявленной хронической фокальной инфекции.
- Больные должны постоянно соблюдать профилактический режим:
- избегать пребывания на солнце, ветру, морозе;
- пользоваться фотозащитными кремами, в солнечные дни - широкополой шляпой или зонтиком.
Больные должны находиться на диспансерном наблюдении дерматолога и ревматолога.
Системная красная волчанка представляет собой прогрессирующее полисиндромное аутоиммунное заболевание с развитием гипериммунного ответа в отношении компонентов собственных клеток (ядерных и цитоплазматических), особенно нативной ДНК, системной дезорганизацией соединительной ткани и генерализованным поражением сосудов.
Болеют преимущественно женщины молодого и среднего возраста, мужчины - в 10 раз реже.
СКВ может провоцироваться: родами, абортами, избыточной инсоляцией, гормональными нарушениями, инфекционными агентами и начинаться с рецидивирующего артрита, лихорадки, недомогания, кожных высыпаний.
В последующем развиваются прогрессирующие патологические изменения в различных органах: полиартрит с артралгиями, миозит с миалгиями, полисерозиты (сухой или выпотной плеврит, перикардит, перитонит), люпус-кардит, синдром Рейно, люпус-нефрит, пневмонит, астеновегетативный синдром, полиневриты, цереброваскулиты с психическими нарушениями, лимфаденопатия, гемолитическая анемия, лейкопения, тромбоцитопения и др. Обнаруживаются LE-клетки и аутоантитела к ДНК.
Поражения кожи при системной форме более разнообразны и распространены, чем при кожной. Иногда (у 10-15% больных) они отсутствуют (lupus sine lupo), однако это состояние носит временный, преходящий характер. Особое диагностическое значение имеет отёчная эритема в средней зоне лица - так называемая волчаночная бабочка. Постепенно эритема может распространяться на шею и грудь. На коже туловища и конечностей могут быть неспецифические полиморфные высыпания: эритематозные и геморрагические пятна, уртикарные элементы, папулы, иногда везикулы и пузыри с геморрагическим содержимым. Типичным проявлением являются так называемые капиляриты: синющные пятна на подушечках пальцев и паронихии. Часто поражается слизистая рта в виде белесоватых группирующихся папул на ярком эритематозном фоне. Нередко возникают пузыри с геморрагическим содержимым, реже встречаются эрозивно-язвенные высыпания. Характерны дистрофические изменения ногтей и диффузная алопеция.
Лечение системной красной волчанки должно быть комплексным:
- адекватная противовоспалительная и иммуносупрессивная терапия
- кортикостероиды,
- циклофосфан,
- биологически активные препараты
- симптоматические и защитные средства.
Прогноз при системной красной волчанке остается в целом неблагоприятным.
Больные должны находиться под диспансерным наблюдением ревматолога.
Склеродермия- аутоиммунное заболевание, которое характеризуется системным поражением соединительной ткани с преобладанием фиброзно-склеротических и сосудистых нарушений, развивающимися приемущественно в коже и подкожно-жировой клетчатке.
Склеродермия является мультифакториальным заболеванием, в основе которого лежит дисрегуляция в соединительной ткани с аутоиммунным механизмом. Выявляются антитела к различным ядерным компонентам: ядерным белкам, ценромерам фибробластов, а так же к коллагену.
Классификация склеродермии. Выделяют ограниченную склеродермию (кожная форма), которая подразделяется на бляшечную, линейную и мелкоочаговую и системную склеродермию, представленную акросклеротическим вариантом (CREST-синдром) и диффузной прогрессирующей склеродермией.
Провоцирующими факторами являются: гормональные расстройства (менопауза, роды, удаление яичников), фокальная инфекция, УФО, переохлажедение, механическая травма, введение сывороток, некоторых лекарств.
Ограниченная склеродермия встречается чаще у женщин в возрасте 40-60 лет. Высыпания на коже могут быть как единичные так и множественные.
Бляшечная склеродермия встречается наиболее часто. Для характерно образование сначала одного или нескольких сиреневых пятен различной величины с четкими границами без субъективных ощущений. Постепенно, в течение месяца происходит уплотнение (склерозирование) пятна в центре с образованием бляшки. По периферии уплотнения не наблюдается, сохраняется лиловый венчик. Очаг может постепенно расти, меняя свой цвет на желтоватый (цвет слоновой кости). Пока существует венчик, заболевание находится в прогрессирующей стадии. Со временем (месяцы и годы) очаг разрешается, формируется участок атрофии. Таким образом, заболевание в своем течении проходит 3 стадии: 1) пятно 2) бляшка 3) атрофия. Локализация процесса - любая (кроме головы).
Полосовидная (линейная) склеродермия наблюдается чаще у детей. Процесс, как правило, представлен одним очагом, распространяющимся линейно, по ходу сосудисто-нервного пучка по длине конечности или с волосистой части головы на лоб и спинку носа, напоминая рубец от удара саблей. Для этой формы типична достаточно глубокая атрофия кожи и подлежащих тканей.

Отличительной особенностью является очень короткий период пятна и бляшки и быстрое возникновение атрофии в виде небольших участков белого цвета (белые пятна). Субъективно часто сопровождается зудом, чего не наблюдается при других формах склеродермии.
Лечение ограниченной склеродермии патогенетическое. При возникновении и обострениях заболевания используется курсовое лечение бензилпенициллином (по 500 т. ЕД. 4 раза в сутки, на курс 28млн ЕД. Эффективны небольшие дозы Д-пенициламина (0,5-0,9 г в сутки, курс 3-6 мес. При мелкоочаговой форме применяют курсы унитиола внутримышечно. Применяют цитостатики.
Акросклеротический вариант системной склеродермии начинается обычно исподволь, чаще всего с поражения кожи пальцев рук. Начальные проявления напоминают симптомы болезни Рейно: больные жалуются на похолодание пальцев, чувство покалывания и ползания мурашек, кожа приобретает цианотичный оттенок. Иногда пальцы белеют и становятся нечувствительными.
Постепенно возникает плотный отек и склерозирование кожи, она становится плотной как дерево, гладкой, блестящей, тесно спаянной с подлежащими тканями. Цвет меняется на желтовато-белый, пальцы неподвижны, слегка согнуты. Постепенно процесс распространяется в проксимальном направлении и доходит до плеча. Обычно через 2-3 года в процесс вовлекается кожа лица. Она уплотняется, приобретает восковидный цвет, лицо становится маскообразным.
Прогрессируя крайне медленно, процесс в дальнейшем или останавливается и идет на разрешение, или может принять характер диффузной склеродермии. Возможно поражение внутренних органов (чаще пищевода в виде его сужения)
Часто в склерозированной коже отмечаются отложения солей кальция, появление телеангиэктазии.
Учитывая описанные выше клинические проявления, акросклеротический вариант склеродермии также обозначается как CREST-синдром (что обозначает характерные клинические проявления: С - кальциноз, R - феномен Рейно, Е - esophagus - поражение пищевода, S - склеродактилия, Т - телеангиэктазии)
Для диффузной склеродермии характерно острое или подострое течение заболевания с быстрым прогрессирующим поражением внутренних органов. Такие пациенты наблюдаются у терапевтов. Поражение кожи диффузное, быстро прогрессирующее. Характерно развитие отёка всей или почти всей кожи. Отёк очень плотный, ямка при надавливании не образуется. Кожа сероватая с синюшным оттенком. Постепенно кожа как бы спаивается с подлежащими тканями. Движения затруднены, мимика ограничена. Развивается атрофия подкожно-жировой клетчатки и мышц. Отмечается синдром Рейно. Поражаются внутренние органы: Лёгкие (пневмофиброз), желудочно-кишечный тракт, прежде всего отмечается сужение пищевода, почки, сердце.
Диагностика. Пациенты с подозрением на системную склеродермию подлежат обследованию у специалистов (дерматолог, терапевт, невропатолог, офтальмолог), лабораторно (характерно повышение СОЭ, С-реактивного белка, уровня глобулинов, обнаружение аутоантител в крови (антиядерные антитела, ревматоидные антитела, антитела против цитоплазматической РНК, антитела против коллагена).и др.), рентгенологически (обследование пищевода (сужение),органов грудной клетки (диффузный пневмосклероз), почек (нефросклероз). рекомендуется проведение эхокардиографии, ЭКГ (выявление диффузных изменений в миокарде), а также исследование сосудистых рефлексов на кистях.
При акросклеротическом варианте склеродермии используют D-пенициламин в высоких дозах в течение нескольких лет. Препарат применяется в связи с его способностью подавлять избыточное фиброобразование и оказывать противовоспалительное действие на соединительную ткань дермы. Препарат используют вместе с витамином В6. Недостатком является большое количество побочных эффектов.
Также применяется унитиол, витамины А, Е, С, Д, вазоактивные препараты, физиотерапевтические процедуры (ультразвук, диадинамические токи, электро- и фонофорез с лидазой), массаж, лечебную гимнастику. Также используют цитостатики.
В стадии уплотнения показаны ферментные препараты (лидаза).
Дерматомиозит- системное прогрессирующее заболевание, характеризуется преимущественным поражением поперечнополосатой и гладкой мускулатуры с нарушением двигательной функции, а также кожи. Различают первичный (идиопатический) и вторичный (симптоматический) дерматомиозит, развивающийся при раке внутренних органов (паранеопластический). Наиболее вероятна иммунная природа заболевания.
Идиопатический дерматомиозит чаще развивается постепенно, характеризуясь общими (субфебрилитет или лихорадка до 38-39°С), мышечными (миалгии, нарастающая мышечная слабость) и кожными проявлениями.
Поражение носит полиморфный характер с преобладанием эритемы, отека, преимущественно на открытых частях тела. Характерны периорбитальный отек и эритема, имеющая лиловый оттенок (симптом "'очков"). Кожный синдром может предшествовать появлению основного признака дерматомиозита - поражению мышц.

Поражение внутренних органов не превалирует в клинической картине болезни. Может отмечаться кальциноз пораженных тканей (синдром Тибьержа-Вейссенбаха).
Диагностические критерии дермтомиозита: типичные кожные проявления; прогрессирующая слабость в симметричных отделах проксимальных мышц конечностей; повышение концентрации сывороточных мышечных ферментов; миопатические изменения при электромиографии; картина полимиозита при биопсии мышц; увеличение креатининурии; уменьшение мышечной слабости при лечении кортикостероидами.
Жизненно важна дифференциация первичного (идиопатического) и вторичного (паранеопластического) дерматомиозита, определяющая тактику лечения и прогноз.
в зависимости от активности и течения процесса при первичном дерматомиозите:
- преднизолон в течение длительного периода с постепенным снижением дозы и корригирующим лечением;
- цитостатики;
- аминохинолиновые препараты (плаквенил, делагил и др.);
- экстракорпоральные методы лечения.
При паранеопластическом дерматомиозите:
Клинические исследования
Отзывы потребителей
Ларун об эмульсии Ла-Кри (irecommend.ru)
«У моего ребёнка периодически возникает аллергический дерматит, который проявляется, как зудящая сухая корочка на коже, от зуда дочь даже просыпалась ночью и плакала. Мы перепробовали несколько разных средств и не одно не давало такого эффекта, как эмульсия Ла-Кри. Это замечательное средство быстро впитывается в кожу, не оставляя не ней эффекта плёнки и прочих ненужных эффектов, когда речь идёт о ребёнке. Уверена мамы меня понимают)
Эмульсия эффективно увлажняет и буквально на глазах, кожа становится значительно приятней на ощупь и на вид тоже. При ежедневном применении, кожа у моего ребёнка становилась всё лучше, быстро зажили маленькие ранки и неровности.
Плюс к этому всему, вполне доступная цена, около 250 руб. При том, что эмульсия Ла-Кри, экономично расходуется и хватает её надолго.
Единственное, что мне не очень нравиться, это аромат. Но почитав другие отзывы, понимаю, что это субъективно. Просто это лично моё ощущение. Но я готова простить производителю этот маленький недостаток, ради производимого на кожу моего ребёночка, эффекта.
И ещё один нюанс, который мне лично очень нравиться, хотя вроде бы и мелочь- это мягкая мембрана, благодаря которой мягко выдавливается небольшое количество эмульсии и вообще создаётся ощущение более дорогого продукта.
Юлия Низовцева о креме Ла-Кри для чувствительной кожи (флап.рф)
«Не боюсь им мазать покраснение у ребенка, и сама часто пользуюсь кремом. В аптечке всегда лежит тюбик на всякий случай, а случаи такие очень часто случаются. Особенно летом, когда жарко, бывают раздражения, потнички из-за жары, крем наношу и все проходит. Пользуюсь им после душа. Наношу совсем немного на кожу и уже через 5 минут ничего не остается, крем легко впитывается и не остается на одежде.
- Ягодка Валентина Степановна, Лекарственные растения в дерматологии и косметологии, изд-во Наукова думка, 1991.
- Молочкова Юлия Владимировна, Дерматология. Краткий справочник, ГЭОТАР-Медиа, 2017.
- Бауманн Лесли, Косметическая дерматология. Принципы и практика, МЕДпресс-информ, 2016.
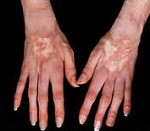
Системная склеродермия - диффузная патология соединительной ткани, для которой характерны фиброзно-склеротические изменения кожи, суставно-мышечного аппарата, внутренних органов и сосудов. Типичными признаками системной склеродермии служат синдром Рейно, уплотнение кожи, маскообразное лицо, телеангиэктазии, полимиозит, суставные контрактуры, изменения со стороны пищевода, сердца, легких, почек. Диагностика системной склеродермии основана на комплексе клинических данных, определении склеродермических аутоантител, биопсии кожи. Лечение включает антифиброзные, противовоспалительные, иммуносупрессивные, сосудистые средства, симптоматическую терапию.
Общие сведения
Причины
Точные представления о причинах системной склеродермии отсутствуют. Накопленные наблюдения позволяют лишь высказывать отдельные этиологические гипотезы. В пользу генетической детерминированности свидетельствуют факты семейной истории системной склеродермии, а также наличие у ближайших родственников других склеродермических болезней, коллагенозов (СКВ, ревматоидного артрита, синдрома Шегрена), микроангиопатий, кардиопатий и нефропатий неизвестного генеза. Выявлена ассоциация склеродермии с определенными антигенами и аллелями HLA-системы, определяющими иммунный ответ, что также свидетельствует о наличии генетического следа в генезе патологии.
Наряду с наследственной теорией, широко обсуждается роль инфекции, в первую очередь цитомегаловирусной. Некоторые пациенты связывают дебют заболевания с перенесенным гриппом или стрептококковой ангиной. Ряд наблюдений указывает на триггерную роль химических агентов: кварцевой и угольной пыли, растворителей, лекарственных средств (в частности, блеомицина и других цитостатиков). Доказано участие вибрационного воздействия, стресса, охлаждения и обморожения, травм в запуске иммунопатологических сдвигов при системной склеродермии. Фоном для развития системного склероза может служить гормональная перестройка, обусловленная пубертатом, родами, абортом, климаксом. У отдельных пациентов началу заболевания предшествуют операции (удаление зуба, тонзиллэктомия и др.) и вакцинация. Т. о., на основании имеющихся данных можно сделать вывод о мультифакториальном генезе системной склеродермии, сочетающем в себе сложное взаимодействие эндо- и экзогенных факторов с наследственной предрасположенностью.
Патогенетические механизмы системной склеродермии изучены лучше этиологии. Ключевую роль в них играют нарушения клеточного и гуморального иммунитета, приводящие к повышению числа CD4 + и В-лимфоцитов, и реакция гиперчувствительности, обусловливающая образование широкого спектра аутоантител (антинуклеарных, антицентромерных, анти-Scl-70, антинейтрофильных, антиэндотелиальных, цитоплазматических, АТ к соединительной ткани и др.) и циркулирующих иммунных комплексов. Подобная иммунная активация способствует гиперактивности фибробластов и повреждению сосудистого эндотелия. Специфика заболевания определяется генерализованным склерозом органов и тканей (кожи, костно-суставной и мышечной системы, ЖКТ, сердца, легких, почек) и развитием облитерирующей микроангиопатии. Рассмотренный механизм позволяет отнести системную склеродермию к аутоиммунным заболеваниям.
Классификация
Системная склеродермия (диффузная или генерализованная склеродермия, прогрессирующий системный склероз) может протекать в нескольких клинических формах:
- Пресклеродермия не имеет дерматологических проявлений и сопровождается только феноменом Рейно.
- Для диффузной склеродермии патогномонично стремительное развитие, поражение кожи, сосудов, мышечно-суставного аппарата и внутренних органов в течение первого года заболевания.
- Лимитированная форма протекает с медленно развивающимися фиброзными изменениями, преимущественным поражением кожных покровов и поздним вовлечением внутренних органов.
- При склеродермии без склеродермы отмечаются только висцеральные и сосудистые синдромы без типичных кожных проявлений.
- Перекрестная форма может проявляться сочетанием системной склеродермии с дерматомиозитом, полимиозитом, СКВ, РА, васкулитами.
Системная склеродермия может протекать в хронической, подострой и острой форме. При хроническом течении на протяжении многих лет единственным указанием на заболевание служит синдром Рейно; другие типичные поражения развиваются постепенно и длительно. При подостром варианте системной склеродермии преобладает кожно-суставной (склеродермия, полиартрит, полимиозит) и висцеральный синдром (сердечно-легочный) при незначительных вазомоторных нарушениях. Острая форма патологии отличается стремительным (в течение 12 мес.) формированием системного фиброза и микрососудистых нарушений. Различают три степени активности системной склеродермии: I – минимальную, типичную для хронического варианта; II – умеренную, обычно встречающуюся при подостром процессе; III – максимальную, сопровождающую течение острой и иногда подострой форм.
Симптомы системной склеродермии
Клиническая специфика системной склеродермии заключается в полиморфности и полисиндромности проявлений. Варианты развития болезни могут варьировать от маловыраженных форм с относительно благоприятным прогнозом до быстропрогрессирующих диффузных поражений с ранним фатальным исходом. В дебюте системной склеродермии, еще до появления специфических поражений, отмечается потеря веса, слабость, субфебрилитет.
Наиболее ранним признаком заболевания служит синдром Рейно, характерный для 99% пациентов и протекающий с преходящими пароксизмами вазоспазма. Под воздействием стресса или охлаждения пальцы рук резко бледнеют, затем кожа приобретает синевато-фиолетовую окраску. Сосудистый спазм может сопровождаться чувством зябкости и онемения кистей. После разрешения вазоконстрикции наступает стадия реактивной гиперемии: кожа становится ярко-розовой, появляется ощущение ломоты и боли в пальцах. Феномена Рейно при склеродермии может носить системный характер, т. е. распространяться на сосуды кожи лица, языка, почек, сердца и др. органов.
Кожный синдром присутствует у большинства больных системной склеродермией. В своей эволюции он проходит 3 фазы: воспалительного отека, уплотнения (индурации) и атрофии кожи. Начальную стадию характеризует появление плотного отека кожи рук и ног, сопровождающегося зудом. В дальнейшем развивается склеродактилия (утолщение кожи пальцев), образуются трофические язвы, деформируются ногти. Лобные и носогубные складки сглаживаются, в результате чего лицо приобретает маскообразное выражение. Вследствие атрофии сальных и потовых желез кожа становится сухой и грубой, лишенной волосяного покрова. Часто обнаруживаются телеангиэктазии, депигментация или гиперпигментация кожи, подкожные кальцинаты.
Мышечно-суставной синдром также часто сопутствует системной склеродермии. Типичны отечность и скованность суставов, артралгии – данный симптомокомплекс носит название склеродермического полиартрита. Вследствие уплотнения кожи формируются сгибательные контрактуры суставов, развиваются теносиновиты. Возможен остеолиз ногтевых фаланг, приводящий к укорочению пальцев. Поражение мышц при системной склеродермии протекает по типу полимиозита или невоспалительной миопатии.
Висцеральные поражения могут затрагивать ЖКТ (90% случаев), легкие (70%), сердце (10%), почки (5%). Со стороны органов пищеварения отмечается дисфагия, изжога, тошнота и рвота. Развивается рефлюкс-эзофагит, усугубляющийся образованием язв и стриктур пищевода. На этом фоне у больных системной склеродермией повышен риск формирования пищевода Барретта и аденокарциномы. При поражении тонкого кишечника возникает диарея, метеоризм, похудание; при вовлечении толстого кишечника – запоры и кишечная непроходимость.
Поражение легких при системной склеродермии может выражаться в виде пневмофиброза и легочной гипертензии. Оба синдрома проявляются непродуктивным кашлем, прогрессирующей экспираторной одышкой и дыхательной недостаточностью. Поражение легких служит ведущей причиной летальных исходов у больных системной склеродермией, поэтому расценивается как прогностически неблагоприятный фактор. При вовлечении сердца могут развиваться аритмии, перикардит (адгезивный или экссудативный), эндокардит, сердечная недостаточность.
Почечный синдром при системной склеродермии чаще протекает в форме латентной нефропатии с умеренными функциональными нарушениями. Однако у ряда больных в первое пятилетие от дебюта заболевания развивается грозное, потенциально летальное осложнение - острая склеродермическая почка, которая протекает с гиперренинемией, злокачественной артериальной гипертензией, тромбоцитопенией и гемолитической анемией, стремительно нарастающей почечной недостаточность. В числе прочих синдромальных проявлений системной склеродермии встречаются полиневропатия, синдром Шегрена, аутоиммунный тиреоидит, первичный билиарный цирроз печени и др.
Диагностика
Американской коллегией ревматологов разработаны критерии, на основании которых может быть выставлен диагноз системной склеродермии. Среди них выделяют большой критерий (проксимальная склеродермия - уплотнение кожи кистей рук, лица и туловища) и малые (склеродактилия, дигитальные рубчики, двусторонний пневмофиброз). При выявлении двух малых или одного большого признака клинический диагноз можно считать подтвержденным. Дифференциально-диагностические мероприятия проводятся как внутри группы склеродермических болезней, так и среди других системных заболеваний: синдрома Шегрена, полимиозита, дерматомиозита, облитерирующего тромбангиита и мн. др.
Общеклинические анализы малоинформативны, а выявляемые в них изменения – неспецифичны. Со стороны крови отмечается гипохромная анемия, лейкоцитопения или лейкоцитоз, умеренное увеличение СОЭ. В общем анализе мочи может выявляться протеинурия, лейкоцитурия, микрогематурия. Биохимические показатели указывают на признаки воспаления (повышение уровня серомукоида и фибриногена, СРБ, РФ). Наибольшее значение имеют результаты иммунологического обследования. При системной склеродермии в крови обнаруживаются склеродермические аутоантитела-маркеры: АТ к Scl-70 и антицентромерные АТ.
Среди инструментальных методик для ранней диагностики системной склеродермии наибольшую ценность представляет капилляроскопия ногтевого ложа, позволяющая выявить начальные признаки болезни. Для оценки состояния костной системы проводится рентгенография кистей. С целью выявления интерстициального пневмофиброза целесообразно выполнение рентгенографии и КТ легких. Для исследования ЖКТ назначают рентгенографию пищевода, рентгенографию пассажа бария по кишечнику. Электрокардиография и ЭхоКГ необходимы для выявления кардиогенных поражений и легочной гипертензии. Электромиография позволяет подтвердить миопатические изменения. Для гистологической верификации системной склеродермии проводится биопсия кожи, мышц, почек, легких, перикарда.
Лечение системной склеродермии
Лицам, страдающим системной склеродермией, следует избегать стрессовых факторов, вибрации, переохлаждения, инсоляции, контакта с бытовыми и производственными химическими агентами, отказаться от курения и употребления кофеина, приема сосудосуживающих средств. Фармакотерапия, ее дозировки и длительность зависят от клинической формы, активности и скорости прогрессирования заболевания, тяжести висцеральных поражений.
Патогенетическая терапия системной склеродермии проводится с использованием сосудистых, антифиброзных и иммуносупрессивных препаратов. Для предупреждения эпизодов сосудистого спазма и профилактики ишемических осложнений назначаются вазодилататоры (нифедипин, верапамил, дилтиазем, циннаризин и др.), антиагреганты (ацетилсалициловую кислоту, пентоксифиллин) и антикоагулянты (гепарин, варфарин). С целью подавления развития системного фиброза используется D-пеницилламин. Противовоспалительная терапия при системной склеродермии включает прием НПВП (ибупрофен, диклофенак, нимесулид) и глюкокортикоидов. Препараты данной группы помогают уменьшить признаки воспаления (миозита, артрита, тендосиновита) и иммунологическую активность. Для замедления прогрессирования системного фиброза может применяться метотрексат, циклоспорин, пульс-терапия циклофосфаном.
Симптоматическая терапия при системной склеродермии направлена на уменьшение расстройств пищеварения, сердечной недостаточности, легочной гипертензии. При развитии склеродермического почечного криза назначается каптоприл, эналаприл; в некоторых случаях может потребоваться проведение гемодиализа. Хирургическое лечение – грудная симпатэктомия - показано при осложненной форме синдрома Рейно.
Прогноз
Прогноз при системной склеродермии в целом неблагоприятный. Самая низкая пятилетняя выживаемость (30-70%) ассоциируется с диффузной формой. Предикторами неблагополучного прогноза выступают легочный и почечный синдромы, дебют болезни у пациентов старше 45 лет. Лимитированная форма и хроническое течение болезни имеют более благоприятный прогноз и лучшую выживаемость, при них возможно планирование беременности и благополучное родоразрешение. Пациенты с системной склеродермией подлежат диспансерному учету и наблюдению каждые 3–6 месяцев.
[youtube.player]Читайте также:
