
Прионы как инфекционные агенты
Несмотря на то, что в наше время медицина развивается с большой скоростью, в ней остается еще много тайн. Одной из таких тайн являются заболевания, вызываемые прионами. Прионы являются инфекционными агентами, устроенными проще, чем вирусы, но при этом способными вызывать инфекционные заболевания, которые на современном этапе развития медицины не поддаются лечению.
Другой инфекционный агент, имеющий крайне простую структуру, это вироиды. Однако для медицины вироиды не представляют особого интереса, так как вызывают заболевания только у растений.
В этом реферате мы подробно рассмотрим свойства и строение и прионов, и вироидов, так как оба этих инфекционных агента являются весьма интересными с точки зрения биологической науки.
Прионы - инфекционные агенты нового типа
В результате обработки PrPSc протеиназой К образуется протеазоустойчивый фрагмент с молекулярной массой 27-30 кДа (молекулярная масса PrP варьирует от 33 до 35 кДа в зависимости от степени гликозилирования). Выявление протеазоустойчивого фрагмента PrPSc с молекулярной массой 27-30 кДа после обработки протеиназой К соскоба ткани миндалевидных желез до сих пор используется при диагностике прионных заболеваний.
На основании имевшихся к 1982 г. экспериментальных данных С. Прузинер сформулировал прионную концепцию. Эта концепция подразумевала следующее:
- инфекционным агентом является белок PrPSc,
- инфекционный агент PrPSc может реплицировать себя в отсутствие нуклеиновой кислоты,
- превращение белка из нормальной формы (PrPC) в инфекционную (PrPSc) происходит путем конформационного перехода,
- конформационный переход PrPC в PrPSc может происходить
спонтанно, приводя к спорадическим формам прионных болезней. Он может быть вызван поступлением в организм патологической формы PrPSc извне (приобретенные формы прионных заболеваний). Наконец, переход может произойти из-за мутаций в гене Prnp, способствующих образованию PrPSc из PrPC (наследственные формы прионных заболеваний).
К настоящему времени концепция прионов получила убедительные экспериментальные подтверждения. Если размножение PrPSc после попадания в организм происходит путем наведения патологической конформации на PrPC, то организмы, лишенные PrPC, должны быть устойчивы к прионной инфекции. Это и было показано с использованием трансгенных мышей, гомозиготных по делеции гена Prnp (Prnp0/0). Введение гомогената мозга мышей, больных скрэйпи, трансгенным мышам Prnp0/0 не приводило к развитию болезни ввиду отсутствия нормального PrPC. Более того, оказалось, что в отсутствие PrPC не происходит не только репликации приона, но и повреждения нервной ткани. PrPC также необходим для транспорта инфекционного агента периферическими нервами к центральной нервной системе.
Окончательное доказательство концепции прионов долгое время сдерживалось невозможностью получения значительного количества PrPres - формы PrPSc, образуемой in vitro, которая устойчива к частичному протеолизу и способна вызывать болезнь при введении экспериментальным животным. Недавно было показано, что фрагмент рекомбинантного PrP мыши с 89 по 231 аминокислоту, экспрессированный в Escherichia coli, образует амилоидные фибриллы in vitro, которые при введении трансгенным мышам, экспрессирующим этот же фрагмент PrP, приводят к развитию неврологической картины прионного заболевания.
Разработка системы циклической амплификации прионной формы
белка PrP, с помощью которой возможно формирование значительного количества PrPres in vitro, позволила получить и продемонстрировать ее инфекционность. Начальной матрицей для образования белка PrPres служил PrPSc - патологический белок из гомогената мозга хомяков, зараженных скрэйпи. Инкубация минимального количества PrPSc (гомогенат мозга хомяков, больных скрэйпи, разводили в 104 раз) с избытком PrPC приводила к образованию агрегатов PrPres. Агрегаты PrPres разрушали ультразвуком на более мелкие, разводили в 10 раз суспензией, содержащей избыток PrPC, и инкубировали снова. В результате много раз повторенного циклического процесса, включающего инкубацию PrPres с PrPC, разрушение агрегатов ультразвуком и последующее разведение, достигалось уменьшение содержания исходного инфекционного агента в реакционной смеси от 104 раз (в первом цикле) до 1055 раз (в заключительном). Образование PrPres в первом цикле происходило на матрице PrPSc, в последующих циклах превращению PrPC в инфекционную форму способствовал PrPres, полученный in vitro. Биохимические и структурные свойства PrPres оказались такими же, как свойства PrPSc, изолированного из мозга больных животных. Интрацеребральное введение PrPres здоровым хомякам индуцировало у них скрэйпи и приводило к смерти. Гистологический анализ мозга умерших животных показал губчатую дегенерацию нервной ткани, неотличимую от таковой у животных, зараженных PrPSc, образованным in vivo. Однако оказалось, что PrPres гораздо менее инфекционен, чем патологический белок, продуцируемый in vivo. Причины таких различий в инфекционности пока не выяснены.
Метод циклической амплификации прионного инфекционного агента эффективен для диагностики губчатых энцефалопатий человека, поскольку позволяет детектировать PrPSc в тканях и биологических жидкостях человека на ранних стадиях развития болезни.
Передача прионной инфекции между видами млекопитающих ограничена межвидовыми барьерами. Губчатые энцефалопатии передаются между особями одного вида или особям близкородственных видов. Например, болезнь Крейцфельда-Якоба передается от человека человеку, и от человека шимпанзе; скрэйпи же передается среди овец и коз, но не передается шимпанзе. Также неизвестны случаи заражения скрэйпи человека. В то же время межвидовые барьеры не абсолютны. Например, возможно заражение хомяков скрэйпи и коз болезнью Крейцфельда-Якоба. Межвидовые барьеры могут выражаться не столько в невозможности передачи инфекции животным отдаленного вида, сколько в удлинении инкубационного периода, а также в том, что заболевают не все, а какая-то часть экспериментально зараженных животных. Считается, что межвидовые барьеры вызваны различиями в первичной структуре PrP у млекопитающих разных видов. Подтверждением этому послужили следующие наблюдения. Трансгенные мыши, экспрессирующие PrP хомяка, оказались высокочувствительны к заражению прионами хомяка в отличие от мышей дикого типа. Передача болезни Крейцфельда-Якоба от человека мыши ограничена межвидовым барьером, однако трансгенные мыши, экспрессирующие PrP человека, подвержены заражению этой болезнью. Впоследствии выяснилось, что передача прионной инфекции ограничена не только отличиями в первичной структуре белка PrP, но и штаммовой принадлежностью приона.

Фото: M24.ru/Евгения Смолянская
За свою историю человечество сталкивалось с огромным количеством войн, эпидемий, стихийных бедствий и других катаклизмов. В XXI веке, когда с эпидемиями, казалось, было покончено, у человечества появился новый вызов – прионы. Что это такое, чем они грозят людям и почему прионами так интересуются ученые всего мира – в материале M24.ru.
Ты помнишь, как все начиналось
В двадцатые годы прошлого столетия врачи столкнулись с новым и неизведанным доселе заболеванием. Немецкий невропатолог Ганс Герхард Крейтцфельдт наблюдал в своей клинике одну пациентку – 20-летнюю девушку. На начальной стадии болезни у нее была нарушена чувствительность в руках и ногах, быстро прогрессировали расстройства памяти, нервной деятельности, больная все чаще впадала в бессознательное состояние. Через несколько месяцев девушка умерла от расстройств дыхания и сердечной деятельности. Невропатолог, который в будущем станет видным нацистским врачом и будет принимать участие в программе "Эвтаназия", задокументировал ход болезни.
Спустя несколько месяцев доктор Альфонс Мария Якоб из Гамбурга столкнулся с тремя аналогичными пациентами. Молодые люди страдали от расстройств нервной деятельности, глотания, практически не осознавали происходящее вокруг и вскоре умерли. При вскрытии Якоб увидел интересное явление, которое раньше врачам наблюдать не приходилось, – поражен у больных был только мозг. Была зафиксирована массовая гибель клеток серого вещества головного мозга, а сохранившиеся нейроны отличались необычным набуханием. Ни в одном другом органе не было зафиксировано никаких патологических изменений. В память о двух первооткрывателях заболевание получило название болезни Крейтцфельдта – Якоба.
В те далекие годы вирусология как наука находилась еще в зачаточной стадии. Поэтому заболеванию было суждено долгое время оставаться в забвении. Этому поспособствовали Великая депрессия и Вторая мировая война. И лишь в пятидесятые годы прошлого века ученые начали активно интересоваться, что же все-таки происходит с людьми, которым не посчастливилось подхватить болезнь Крейтцфельдта – Якоба.
В то же время ученые открывают еще два заболевания, которые по своим симптомам и течению весьма и весьма напоминают описанный выше страшный недуг – куру и скрейпи. Первая болезнь была распространена среди народности форе на острове Папуа – Новая Гвинея, а вторым страдали овцы по всему миру. Но важным оказалось другое: симптомы болезней несколько отличались от болезни Крейтцфельдта – Якоба, но характер поражений был практически идентичен – образование пустот в тканях головного мозга и массовая гибель нервных клеток.
Казалось бы, все ясно. Имеется болезнь, ее вызывает какой-то вирус или бактерия, давайте разберемся, кто является возбудителем и устраним причину. Но не тут-то было! Все оказалось не так просто.

"Познавательные фильмы": Вакцины
Исследования
Ученым удалось достаточно быстро установить, почему болеют папуасы. Выяснилось, что заболевают только те из них, кто участвовал в ритуальном поедании тел погибших от куру родственников. Согласно местным верованиям того времени, дети должны были обязательно отведать мозга умершего, считалось, что от этого у них прибавится ума. Неизвестно, прибавлялось ли у детей от этого ума, но все малолетние, участвовавшие в таких трапезах, обязательно оказывались зараженными куру.
Особенно масштабные исследования развернулись с агентом скрейпи. Для начала определили его размеры, они оказались стандартными для вирусов – 17–27 нанометров. После этого вирусологи всего мира стали разбираться в свойствах неизвестного возбудителя заболевания, и тут их ждали сюрпризы. Оказалось, что инфекционный агент совершенно невосприимчив к формалину, пепсину и трипсину, не реагирует на ферменты, разрушающие ДНК и РНК, устойчив к кипячению, ультрафиолетовому излучению и. проникающей радиации! С такими вирусами ученым сталкиваться еще не приходилось.

Фото: M24.ru/Александр Авилов
Больше того, возбудителя заболевания никак не удавалось увидеть в электронный микроскоп, что было уж совсем странно. В то время ученые уже умели распознавать вирусные частицы намного мельче, чем 17 нанометров, но вирус скрейпи (почесуха) так никто и не увидел – наблюдали лишь фрагменты клеточных мембран.
Еще одной интересной загадкой оказалось всякое отсутствие иммунного ответа организма больных. Организм людей, больных куру, и овец, страдавших от скрейпи, никак не реагировал на течение заболевания. При обычных болезнях, вроде гриппа и простуды, в организме увеличивается синтез интерферона (отвечает за иммунитет), что ведет к быстрому выпуску антител, которые соединяются с вирусными частицами и растворяют их. Ученые пытались обнаружить признаки хоть каких-либо антител, но потерпели неудачу.
Отчаявшиеся исследователи начали выдвигать гипотезы, что возбудителем является не вирус, а молекула полисахарида или же белка, но подтверждения эта версия так и не нашла. Ученые топтались на месте, пока в 1982 году американский невролог Стэнли Прузинер не заявил об открытии нового класса инфекционных агентов – прионах.
Что такое прион
До открытия прионов считалось, что болезни человека и животных могут вызываться исключительно живыми организмами или хотя бы вирусами, содержащими нуклеиновую кислоту. Однако все оказалось не так просто. Прион – это особый вид белка, который присутствует в любом человеческом организме.
Выяснилось, что либо под воздействием непонятных факторов, либо из-за мутаций в организме некоторых людей нормальный прионный белок, входящий в состав клеточных мембран, заменяется "неправильным". Второй вид прионного белка имеет другую структуру, вызывает гибель клеток, но самое интересное – способен самостоятельно размножаться (без каких-либо ДНК и РНК!) и менять нормальные прионы в соседних клетках на дефектные.

"Познавательный фильм": Вирусы и защита от эпидемий
Таким образом, прионы оказались единственным видом инфекционных агентов, которых никак нельзя причислить к живым существам. Ведь, по своей сути, они не содержат никакой генетической информации и самостоятельно синтезируются организмом.
Естественно, исследователей заинтересовал самый главный вопрос – а зачем вообще в человеческом организме нужны прионы? В настоящее время известно уже достаточно много прионных болезней. Все они являются экстремально редкими, самая распространенная – болезнь Крейтцфельдта – Якоба – наблюдается у одного из миллиона человек. Также известно о синдроме Герстманна – Штраусслера – Шайнкера, фатальной семейной бессонице и куру. Некоторые исследователи включают в группу прионных заболеваний человека также болезнь Альперса у детей, амиотрофический лейкоспонгиоз (описан белорусскими учеными в конце прошлого века, болели работники одной из ферм) и спонгиоформный миозит (мышечное истощение).
Все эти заболевания являются смертельными, и лекарств от них пока не предложено. Но все же зачем организм синтезирует прионы? Какую он отводит роль для них?
Зачем нужны прионы?
В 70-е годы прошлого века два английских исследователя – Паттисон и Джебет – изучали на мышах действие вещества под названием купризон. В нормальных условиях оно связывает в организме ионы меди. Животным включили купризон в обязательную диету с целью посмотреть, какое действие он произведет на грызунов. И поразились! После 30 с лишним дней купризоновой диеты совершенно здоровые до этого мыши превратились в тяжелобольных. Причем все признаки заболевания полностью отвечали симптомами скрепи. Часть мышей, участвовавших в эксперименте, вскрыли и посмотрели – оказалось, что в головном мозгу животных произошли абсолютно те же изменения, что и при прионных болезнях.
Возник вопрос: а что если купризон мышам больше не давать? Попробовали – и через несколько дней грызуны выздоровели. Уже через 30 дней у них исчезли и вызванные купризоном изменения в мозговой ткани.
Спустя много лет было выяснено, что прионы весьма и весьма похожи на положительно заряженные частицы двухвалентной меди. И изменения, которые они вызывают в организме, практически идентичны. Таким образом, исследователи сделали вывод о том, что в нормальном состоянии прионы отвечают за оборот металлов, в частности меди. Но эти данные пока остаются лишь гипотезой.

Фото: ТАСС/Станислав Красильников
Еще одна группа американских исследователей принялась копать в другом направлении. Им удалось получить данные, что прионы помогают клеткам мозга прикрепляться друг к другу и участвуют в передаче сигналов внутри клетки. Это означает, что отсутствие прионов или их дефекты не позволяют клеткам мозга получать сигнал о других клеток, что ведет к развитию тяжелых нарушений в работе нервной и других систем организма.
Но самым интересным является предположение о том, что прионы участвуют в механизмах клеточного старения. Не секрет, что долгое время прионные болезни относили к группе старческих болезней, потому что вызываемые ими изменения весьма сходны с другими заболеваниями (вроде болезни Пика, Альцгеймера и других неврологических недугов). Наличие прионной инфекции как бы подталкивает организм к ускоренному старению. Естественно, это ставит очень важный вопрос: если лекарство от таких болезней будет найдено, не станет ли оно своеобразным ключом к долголетию или даже бессмертию организма? Но ответ на этот вопрос пока дать невозможно, поскольку функции прионов изучены еще недостаточно хорошо.
Способы заражения
В заключение поговорим о способах заражения. Их четыре. В первом и самом распространенном случае заболевание возникает как бы из ниоткуда. То есть жил себе человек, да вдруг взял и заболел. Этот путь возникновения болезни называется спорадическим и, кстати сказать, является наиболее распространенным. По нынешним представлениям, это происходит спонтанно под действием каких-то пока не установленных факторов.
Второй способ – наследственный. Некоторые виды болезней являются семейными и возникают из-за мутаций. В свою очередь, гены передаются потомству. Известно около 40 семей, страдающих фатальной бессоницей. Каждый десятый страдающий болезнью Крейтцфельдта – Якоба – страдает семейной формой этого заболевания.
Фото: M24.ru/Михаил Сипко
Третий способ – ятрогенный. Это означает, что заражение прионами произошло по вине медицинских работников при проведении каких-либо оперативных вмешательств. Однако описаны лишь несколько таких случаев, и все они произошло в 70-е годы прошлого века, когда о свойствах прионов еще никто не знал. Так, одна женщина заболела после того, как ей пересадили роговицу глаза от страдавшего болезнью Крейтцфельдта – Якоба мужчины.
А вот последний способ наиболее коварен и опасен. Дело в том, что человек восприимчив к прионам, которые поражают крупный рогатый скот. И при употреблении в пищу мяса больных животных заболевают и люди – у них развивается болезнь Крейтцфельдта – Якоба. В девяностые годы прошлого века настоящая эпидемия этого страдания разразилась в Англии.
Лечения пока нет. Однако ученые уже выяснили, что некоторые виды прионов разлагаются лишайниками, другим удалось описать особые антиприонные антитела (к инфекционным прионам).
Иными словами, перед исследователями стоит весьма непростая задача, которая не только поможет найти лекарство от тяжелых заболеваний, но и, возможно, поможет открыть секрет долголетия. Для этого нужно только одно – понять прионы.
Прионная болезнь – это тяжелый вид нейродегенеративного заболевания у млекопитающих. Его вызывают прионы – возбудители. Эти инфекции поражают живой организм, и впоследствии он умирает.
Что такое прионы
Прионы – это некие белки, они могут содержатся в организме млекопитающих и являются безвредными. Также встречаются и патологические, т.е. вызывающие болезни. Прионы не содержат ничего, кроме белков, и при этом они имеют способность размножаться. Это открытие перевернуло все представления ученых.
Как только белковые прионы попадают в живой организм, они размножаются посредством превращения здоровых прионов в патологические. Для этого процесса требуется много времени, поэтому до развития болезни могут пройти годы.
Свойства прионов
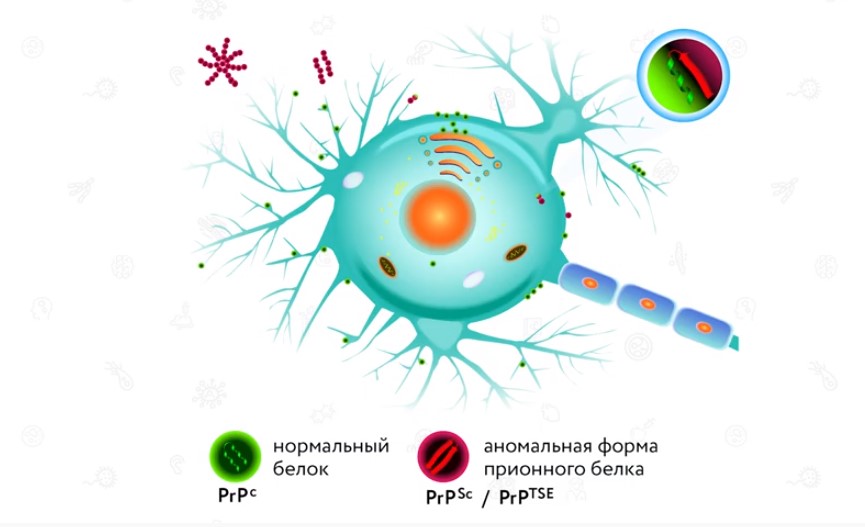
Прионы и вироиды – очень устойчивые соединения. Прион:
- выдерживает кипячение до 3 часов;
- способен выживать при температуре -40 градусов на протяжении нескольких лет;
- защищен от ультрафиолета;
- после обработки формалином не инактивируется.
Из-за особой структуры этих белков живой организм не может с ними бороться, т.е. он не выделяет антитела, фактически не замечает прионы. Это приводит к летальному исходу живого организма.
Что вызывают в организме прионы
Прионные болезни поражают нервную систему человека:
Эти процессы идут без болевых ощущений для зараженного.
Симптомы прионной инфекции
В зависимости от вида заболевания, прионная инфекция может вызывать:
- частые головокружения;
- появление неконтролируемых движения;
- потеря возможности передвигаться и говорить;
- приобретенное слабоумие;
- неконтролируемый смех;
- частичная, а затем и полная потеря сна.
На конечных стадиях заболевания пациент неизбежно погибает.
Как можно заразиться прионами
Прионные заболевания передаются 3 способами:
- Трансмиссивный. Прионные болезни вызываются при передаче белка между млекопитающими. Т.е. человек может заразиться инфекцией от животного. Причины появления инфекции: употребление мяса больной птицы или скота, использование лекарств из крови (лучше заменить на синтетические).
- Наследственный. Развивается из-за генетической мутации, произошедшей в 20-й хромосоме. В этой хромосоме есть участок, отвечающий за безвредный белок. При генных изменениях синтезируется аномальный белок, который и приводит к болезни.
- Спорадический. Спонтанное появление патологического белка.
Какие болезни являются прионными у человека
Сейчас есть 5 прионных болезней. Они атакуют нервную систему, что в итоге приводит к смерти.
Сокращенно ее называют БКЯ. В биохимии – это прогрессирующее поражение коры спинного и головного мозга, а также базальных ганглиев. БКЯ — распространенная прионная болезнь. Заболевание не лечится. Оно может поразить человека любой расы, возраста и пола.
Это наследственный тип прионной болезни, который встречается у 1 из 10 млн человек. Обычно заболевание начинает проявляться у людей в возрасте 40 лет.
Сначала у зараженного происходят частые головокружения и нарушения координации во время ходьбы из-за патологических белков. После человек чаще прихрамывает и теряет способность передвигаться самостоятельно. Параллельно заболевание приводит к глухоте, ухудшению зрения, а также к нарушениям с произведением звуков.
Ученые впервые встретились с этой наследственной болезнью в 1986 году. Случаи заражения инфекцией бывают редко. К 2003 году зарегистрировано 26 семей с такой болезнью. Симптомы могут проявиться у пациента от 25 до 70 лет. Живут после появления признаков болезни от полугода до 4 лет.
Больной утрачивает способность спать. Его мучают постоянные бессонницы. Также наблюдаются нарушения опорно-двигательного аппарата, дрожание, и слабость в мышцах. Из-за отсутствия сна организм пациента полностью истощается.
Этот вид прионной инфекции смогли изучить из-за племени каннибалов из Папу-Новой Гвинеи. До середины прошлого века среди местного населения существовали обычаи поедания мозгов погибших сородичей. Считается, что у 1 из жителей племени развилась прионная инфекция, которая после акта каннибализма передалась его нескольким родственникам.
Это наследственная прионная болезнь, проявляющаяся у людей моложе 18 лет. Инфекции атакуют нервную систему и зрение человека. Зараженного сопровождают эпилептические припадки. Болезнь поражает печень, у пациента развивается гепатит. Человек живет короткий период времени (до 1 года) после появления симптомов болезни из-за печеночной недостаточности.
Диагностика клеток
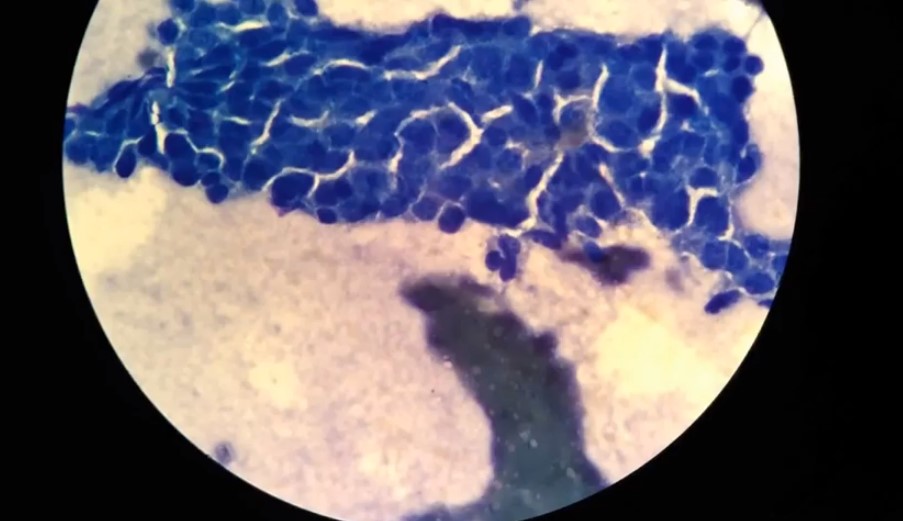
Зачастую обнаружить прионные инфекции у человека удается, когда начинают проявляться основные симптомы. К примеру, нарушение координации из-за потери клеток Пуркинье. Нередко диагноз удается поставить только после смерти пациента, т.к. распространены случаи спонтанного заражения.
Определить наличие прионной болезни можно с помощью диагностики клеток. Ее проводят в специальных лабораториях, и целью ученых является выявление мутации в 20-й хромосоме. Здесь расположен PrPC – ген, отвечающий за наличие нормальных прионов, который при мутации синтезирует аномальный белок.
Лаборатории, способные провести диагностику клеток, преимущественно расположены за рубежом. Но с помощью этих анализов человек получит точный ответ, есть ли в его организме прионные инфекции.
По указу Роспотребнадзора зараженных прионными болезнями нужно известить в течение 2 часов после обнаружения болезни. При этом в больнице необходимо утилизировать все оборудование, с которым больной был в контакте. Больницы не хотят нести большие убытки из-за утилизации дорогостоящего оборудования (к примеру, аппарат МРТ).
Лечение прионных заболеваний

К сожалению, ученые еще не придумали способ вылечить ряд заболеваний, вызванных прионами. Пациентам проводят симптоматическую терапию, чтобы облегчить их боль, но на результат прогноза препараты никак не повлияют. В конечном счете человеческий организм погибает.
в 2019 году ученые из США совершили прорыв в области лечения прионных инфекций. Они смогли замедлить ход заболевания у подопытных мышей с помощью коротких молекул РНК. Подробнее в статье Найдено лекарство для борьбы с инфекциями мозга.
Как уберечься от прионных инфекций
Нет способов обезопасить себя от развития болезней, вызванных прионами. Теоретически человек может пройти диагностику клеток и выяснить, есть ли у него генетические нарушения и предрасположенность к прионной болезни. Но лаборатории, способные провести такие анализы в основном расположены за рубежом, в высокоразвитых странах.

Впервые о возникновении этого нБКЯ предположили в 1996 году. Дальнейшие исследования подтвердили опасения ученых. Возможно, больные ели мясо, в котором содержались прионы мозга коров.
нБКНЯ не имеет конкретного возраста заражения. Инфекция вызывает у зараженных и психологические изменения (нередко это расстройство по ошибке диагностируют как психическое, вместо неврологического). Развитие болезни длится 6-8 месяцев, но иногда может достигать и полутора лет, поэтому врачам сложно поставить правильный диагноз после первых симптомов. Обычно новую форму диагностируют, когда больной перестает контролировать движения.
Последняя стадия нБКЯ проходит с быстрой потерей мышечного контроля. Пациент теряет возможность двигаться и говорить (акинетический мутизм).

Деменции с быстрым прогрессированием могут привести к смерти через несколько дней или месяцев с начала своего проявления. Примерно у 47% больных со спонтанной формой БКЯ обнаружена быстро прогрессирующая деменция, с генетической – 13%, а иные случаи относят к остальным деменциям.
Прионный белок может развиться как спонтанно, так и генетически, поэтому практически все пациенты долгое время об этом не знают.
Окончательный диагноз быстро прогрессирующей деменции ставится после дифференциальной диагностики. Но даже при тщательном обследовании больных некоторые диагнозы ставятся только после вскрытия.

Эта болезнь не позволяет человеку спать, и лишает его возможности выполнять привычные функции. Есть спорадическая и генетическая формы. Генетическая приводит к мутации PrP белка и его преобразование в прионный. Спорадическая форма обнаруживается случайно, без наличия предпосылок. Это заболевание поражает таламус (отдел мозга, отвечающий за сон), поэтому зараженный теряет способность спать.
В среднем, первые симптомы могут появиться у пациента в 40 лет. Поначалу у больного появляются проблемы со сном и судороги. Также во время сна зараженный может непроизвольно двигать конечностями. В результате пациент вообще теряет способность спать, вследствие чего понижается активность мозга и теряется координация мышц.
Для точного диагноза генетической формы нужно провести соответствующие тестирования. Спорадическую форму можно диагностировать по нарушениям в структуре сна. В среднем, человек живет до 3 лет с момента проявления начальных симптомов.
Это заболевание впервые описали более 80 лет назад. В основном синдром развивается у людей среднего возраста (от 40 до 50 лет), и он вызывается мутацией на 20 хромосоме.
Симптомы заболевания примерно такие же, как со спонтанной формой БКЯ. Но деменция прогрессирует годы. Этот синдром может длиться от нескольких месяцев до нескольких лет. В среднем, пациенты живут 5 лет. Заболевание обнаруживается с помощью МРТ.
- 13732
- 11,8
- 21
- 4
Путь прионов

Спонсор конкурса — дальновидная компания Thermo Fisher Scientific.
Биологическая сущность прионов
Рисунок 1. Метафора нейродегенеративного поражения мозга — это губка, в которую превращается нервная ткань в результате массовой гибели нейронов.
И тогда происходит удивительное событие: нормальные молекулы белка, контактируя с патологическими, сами превращаются в них, изменяя свою пространственную структуру (механизм трансформации остаётся загадкой и по сей день) [1]. Таким образом прион, как самый настоящий инфекционный агент, заражает нормальные молекулы, запуская цепную реакцию, разрушительную для клетки.
Некоторые сведения о прионах
Условия возникновения заболеваний
Условия возникновения прионовых болезней уникальны. Они могут формироваться по трём сценариям: как инфекционные, спорадические и наследственные поражения. В последнем варианте главную роль играет генетическая предрасположенность [2].
В последнее десятилетие интерес к этой теме возобновился в связи с возможностью развития диагностики и эффективной терапии [5]. Появилось множество различных объяснений для возрастных нейродегенеративных болезней, — например, окислительная модификация ДНК, липидов и/или белков; соматические мутации; измененный врождённый иммунитет; экзогенные токсины; несоответствия ДНК—РНК; нарушение работы шаперонов; отсутствие одного из аллелей гена [5]. Альтернативным комплексным разъяснением служит то, что различные группы белков могут формировать прионы. Несмотря на то, что небольшое количество прионов может быть удалено посредством путей белковой деградации, их чрезмерное накопление с течением времени позволяет прионам самостоятельно распространяться в организме (рис. 2), что приводит к нарушению деятельности центральной нервной системы [5].

Группы риска прионных заболеваний
Вот кого прионные заболевания могут настичь с наибольшей вероятностью:
- работники пищевой промышленности;
- ветеринары;
- патологоанатомы;
- хирурги;
- пациенты трансплантолога;
- каннибалы;
- лица, в семье которых были замечены синдромы Герстманна—Штрейслера—Шейнклера или фатальной инсомнии.
Лабораторная диагностика и лечение
Диагностика базируется на внутримозговом заражении мышат или хомяков, у которых медленно (до 150 дней) развивается соответствующее заболевание, если пациент был болен [2]. Часто проводится гистологическое исследование головного мозга погибших животных [2].
К сожалению, до настоящего времени еще не разработаны эффективные методы лечения прионовых болезней, хотя попытки предотвратить конформационный переход нормального белка в аномальный производятся. Поэтому самым надёжным способом предупреждения развития инфекционных форм является профилактика [2].
Перспективы
По-видимому, интерес к прионам не угаснет до тех пор, пока предположения на их счёт полностью не подтвердятся и не будут найдены эффективные способы лечения прионных заболеваний. В статье [6] говорится о необходимости современного исследования, которое требует тщательного рассмотрения чужеродных прионов в экстраневрональных тканях.
В качестве модельных объектов авторы использовали мышей: две линии, которые трансгенно экспрессировали овечий прионный белок, и одну линию, которая экспрессировала человеческий прионный белок (рис. 3). Задачей было сравнить эффективность межвидовой передачи инфекции посредством тканей мозга и селезёнки. Внутримозговое заражение чужеродным прионным белком выражалось в отсутствии или небольшом количестве инфекционного агента в мозгах этих мышей. Однако инфекционные чужеродные прионы обнаруживались в селезёнке на более ранних этапах заражения в сравнении с моментом, когда были использованы нейротропные прионы, тем самым определяя, что лимфатическая ткань может быть более пермиссивной к распространению чужеродных прионов по сравнению с мозгом.
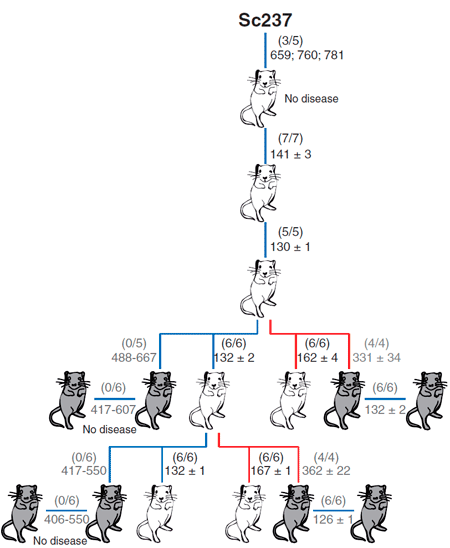
Рисунок 3. Способность приона хомяков Sc237 заражать и передаваться при введении в мозг или селезенку трансгенным мышам, имеющим прионный белок PrP овцы (tg338; белые мыши) или человека (tg7; серые мыши). Число заболевших/инъецированных мышей показано в скобках; ниже приведено среднее время жизни (в днях).
Чем вызвана эта предпочтительная репликация прионов в лимфатических тканях, пока неизвестно. Однако полученные данные показывают, что человек может быть более чувствительным к чужеродным прионам, чем предполагалось ранее на основании присутствия прионов в мозгу, и по этой причине бессимптомный переносчик прионной болезни может быть не распознан. Это ещё раз подтверждает, что такая могущественная биомолекула как прион таит в себе немало загадок, раскрытие которых, возможно, поможет в понимании ряда неразрешимых проблем человечества.
Читайте также:
