
Что такое армянская лихорадка болезнь
Периодическая болезнь является одним из наиболее странных и труднодиагностируемых проблем в здоровье человека. Как правило, больной успевает пройти лечение у врачей практически всех специализаций, не дающее никаких положительных результатов, пока в итоге ни узнает о диагнозе, который доставляет невыносимые страдания.
Описание патологии
Периодическая болезнь знакома человечеству под различными названиями, такими как рецидивирующий полисерозит, средиземноморская лихорадка или армянская болезнь. Это заболевание имеет отношение к наследственным аутосомно-рецессивным дисфункциям и широко распространено в основном среди жителей средиземноморского района планеты. Поэтому зачастую недуг можно встретить у греков, армян, евреев-сефардов, турков и среди многочисленных народов Кавказа.

Клиническая картина периодической армянской болезни
Выделяют следующие разновидности периодической болезни, которые зависят от их локализации:
- Торакальная.
- Лихорадочная.
- Абдоминальная.
- Суставная.
Торакальный тип
Торакальный тип сопровождается процессом воспаления плевры, которое как бы кочует по различным частям грудной клетки. Результаты исследований и жалобы больного, как правило, указывают на сухой плеврит что, безусловно, не может соответствовать действительности. Обострения могут пропасть уже спустя неделю спонтанно.
Абдоминальный вариант
На фоне абдоминального варианта армянской периодической болезни врачи могут заподозрить аппендицит, так как пациент страдает от сильных, даже острых болей в районе живота. Формируется видимость, будто речь идет о непроходимости тонкой кишки либо о холецистите. Зачастую подобные симптомы уже спустя два-четыре дня загадочным образом пропадают. Периодическую болезнь принято диагностировать, прибегая к рентгенологическому обследованию пораженных органов района брюшной полости.

Лихорадочный и суставной виды
На почве лихорадочного вида болезни у пациента резко повышается температура, а течение самого недуга больше напоминает малярийную лихорадку.
Но самым неприятным считается суставной вариант, который проявляется в виде рецидивирующего синовита, а кроме того, моноартрита и артралгии. В том случае, когда артриты задерживаются, существует вероятность появления преходящего остеопороза.
Причины и особенности возникновения
Основная причина армянской болезни кроется в наследственности нарушения вещественного обмена, которые могут сопровождаться увеличением проницаемости сосудов, развитием соединительных тканей, а также склонностью к экссудации или, выражаясь более понятным языком, к отечности. Продолжительное время заболевание способно протекать бессистемно, не вызывая при этом никаких обострений. Но из-за влияния пока еще не изученного комплекса внутренних и внешних предпосылок формируется доброкачественная опухоль в отношении серозных оболочек при средиземноморской лихорадке (армянской болезни).
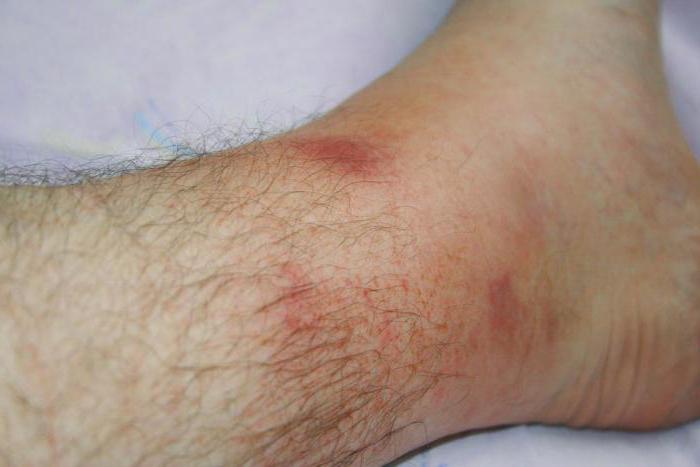
Начало лихорадки сопровождается болевым синдромом. Происходит отложение белка - амилоида в тканях и органах, преимущественно в почках. Атака может растягиваться на несколько дней, после чего состояние больного заметно улучшается ровно до возникновения следующего приступа. Как правило, ремиссия составляет приблизительно три-семь дней.
Отложение вещества амилоида после каждого случая атаки вызывает за собой нарастающее угнетение почек. Хроническая почечная недостаточность впоследствии возникает у двадцати пяти-сорока процентов больных.
Проявления и признаки периодической болезни
Диагностика армянской болезни, как правило, вызывает большие затруднения. Но тем не менее основные характерные черты этого недуга очертить все же можно:
- Лихорадка обычно сопровождает основные этапы обострений. Она идентична по своей типологии малярии и определяется внезапным температурным скачком до сорокоградусной отметки.
- Перитонит наблюдается в девяноста пяти процентах случаев. На фоне воспаления брюшины больных госпитализируют и направляют в хирургические отделения.
- Артрит выступает в роли суставного типа заболевания, которое прослеживается в восьмидесяти процентах случаев.
- К торакальной форме причисляют различные проблемы с дыханием, бронхиты и плевриты, что встречается в шестидесяти процентах ситуаций.
- Комбинированные формы, характеризующиеся значительным увеличением селезенки, поражением лимфоузлов, а также кожной сыпью, которая отдаленно напоминает рожу, встречаются среди пятидесяти процентов. Редко, но бывает, что возникает асептический менингит.

Диагностика периодической болезни
Симптомы армянской болезни (что это, мы пояснили) довольно неприятны.
При диагностировании этого заболевания следует обращать внимание на следующие критерии:
- Периоды коротких атак, которые не связаны с провоцирующим фактором и являются стереотипными.
- Примечательно, что болезнь поражает представителей конкретных этнических групп. При этом у детей и подростков она возникает довольно рано.
- Аналогичные заболевания происходят и у близких родственников.
- Происходит амилоидоз почек, на фоне которого специфику лабораторных показателей крайне сложно определить.
Армянская генетическая болезнь абсолютно несовместима с беременностью. Как бы там ни было, но частота возникновения припадков у будущих мам заметно снижается.

Лечение
Что касается прогнозов, то болезнь может повлечь за собой нетрудоспособность временного характера. При стабильном укреплении амилоидоза вероятно наступление стадии почечной недостаточности, что, скорее всего, неизбежно приведет к инвалидности. Положительный эффект, как правило, наступает тогда, когда лечение начинают своевременно. По этой причине приветствуется диспансерное наблюдение, поэтому доктора его настоятельно рекомендуют страдающим пациентам.
Существует лекарство от армянской болезни. Об этом далее.


Факторы риска проявления болезни
Причиной этого заболевания довольно часто становится психологическая травма. В большинстве своих случаев недуг может обостряться в ситуациях, когда человек оказывается в жизненной ситуации, сопровождающейся длительное время угнетенным и подавленным состоянием, а также ощущением незащищенности. Большое влияние на развитие болезни оказывают пережитые страхи. Случающиеся в раннем возрасте приступы зачастую могут быть спровоцированы прерыванием связи ребенка с матерью. Это объясняет, почему у подавляющего большинства пациентов имеются в наличии все признаки депрессии, в особенности маскированного характера, когда они выражаются не в психических проявлениях, а в болях в различных частях тела, например, в голове, мышцах, суставах либо брюшной полости.
В двадцатом веке у армянского народа существовало огромное число факторов, которые способствовали распространению этого заболевания и увеличению числа соответствующих пациентов. Среди них: переселение, геноцид, разрушение прежних устоев, митинги и землетрясение, а также резня в Сумгаите, война и блокада.
На сегодняшний день во всем мире медициной рассматривается психосоматическое расстройство техногенного характера, которое, как правило, возникает у людей, перенесших различные катастрофы и ужасы. Описание данных факторов и выражает суть изначального формирования периодической армянской болезни. То есть, если говорить о роли, которую играет в развитии этого опасного недуга наследственность, то следует помнить, что это заболевание носит глубинный генетический страх ряда упомянутых народов.
Семейная средиземноморская лихорадка: причины, диагностика, лечение.

Семейная средиземноморская лихорадка (другие названия: периодическая болезнь, армянская болезнь, ереванская болезнь, наследственный семейный амилоидоз без невропатии, пароксизмальный синдром Джэйнуэя — Мозенталя, периодический перитонит, синдром Рейманна, болезнь Сигала — Маму, FTF).
Семейная средиземноморская лихорадка, как и большинство редких болезней, это сложно диагностируемое заболевание. Пациент проходит годы наблюдения у врачей разных специальностей, прежде чем узнает об этом трудном орфанном диагнозе, чаще всего связанном с его национальностью.
Семейная средиземноморская лихорадка. Что это такое?
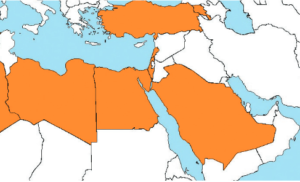
Семейная средиземноморская лихорадка — это наследственное аутосомно-рецессивное заболевание, которое встречается в основном среди жителей средиземноморья. В этом регионе болезнь не является такой уж редкой и составляет 1 случай на 200 человек. Патология часто проявляется у греков, арабов, евреев-сефардов(неашкеназского происхождения), турков и среди многочисленных народов Кавказа. Особенно можно выделить армян. Среди них 15-20% имеют гены, предрасположенные к этой болезни, и если эти гены получены от обоих родителей, начинаются приступы. В западных странах, в том числе и в России, является орфанной болезнью, встречается с частотой 2,5 случая на 100 000 человек.
Семейная средиземноморская лихорадка. Этиология и патогенез.
Семейная средиземноморская лихорадка. Клинические проявления.
Выделяют следующие разновидности семейной средиземноморской лихорадки, которые зависят от их локализации: Торакальная. Лихорадочная. Абдоминальная. Суставная.
Торакальный тип

Торакальный тип возникает у 20-40% пациентов, он сопровождается процессом воспаления плевры. Боль в грудной клетке чаще всего является односторонней. Она может быть настолько сильной , что пациент не может глубоко дышать. Жалобы больного могут принять за сухой плеврит, который самостоятельно проходит без какого-либо вмешательсва примерно через неделю.
Абдоминальный вариант

Как это понятно из названия, пациента с семейной средиземноморской лихорадкой начинает лихорадить. Появляется необыкновенно сильный озноб, резко повышается температура до 39—41°. Лихорадка — наиболее частый и постоянный симптом, встречается в 96–100% случаев. Изначально, резкое повышение температуры принимают за ОРВИ, но антибиотики с температурой не справляются, а сама температура благополучно исчезает через несколько дней вместе с другими симптомами.
Суставной вариант
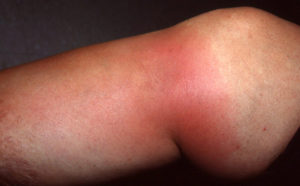
Проявляется в виде поражения суставов (35–80% случаев) в виде рецидивирующего синовита, а кроме того, моноартрита и артралгии. Иногда, это самый первый признак заболевания. Боль может появиться в суставе внезапно, конечность отекает, она гиперемирована. В том случае, когда артриты задерживаются, существует вероятность появления преходящего остеопороза. Суставной вариант — наиболее длительный из всех проявлений семейной средиземноморской лихорадки, боли в суставах могут сохраняться до месяца.
Семейная средиземноморская лихорадка. Причины.
Основная причина семейной средиземноморской лихорадки заключается в наследственном изменении обмена веществ, который иногда провоцирует большую проницаемость сосудов, развитие соединительных тканей, а также склонность к отечности. Болезнь длительное время может протекать бессимптомно, но под воздействием провоцирующих факторов переходит в острую стадию — формируется доброкачественная опухоль в отношении серозных тканях (они образуют наружные оболочки большинства органов и сосудов человека).
Боль — первый предвестник семейной средиземноморской лихорадки. Это происходит отложение белка — амилоида в тканях и органах, преимущественно в почках. Атака лихорадки длится несколько дней, а после бесследно проходит вплоть до следующего приступа. Между приступами проходит примерно три-семь дней.
Отложение вещества амилоида после каждого случая атаки вызывает за собой нарастающее угнетение почек. Хроническая почечная недостаточность впоследствии возникает у 25-40% больных.
Семейная средиземноморская лихорадка. Проявления болезни.
Впервые столкнувшись с проявлениями семейной средиземноморской лихорадки врачам очень сложно идентифицировать именно это состояние, но с течением времени периодичность симптомов, спады и падения обострений могут навести на мысль об этой редкой патологии. Первое, что нужно выделить — это лихорадка, она встречается почти в 100% обострений, внезапный скачок температуры выше 40 градусов должен насторожить врача. Острая боль из-за перитонита наблюдается в 95% случаев, таких пациентов наблюдают хирурги. В 80% случаев болезнь будет проявляться болью в суставах из-за развивающегося артрита. 60% больных испытывают проблемы с легкими: их поражают бронхиты и плевриты. 50% пациентов имеют смешанную форму проявления заболевания, она характеризуется спленомегалией (увеличением селезенки), поражением лимфоузлов и кожной сыпью. В отдельных случаях развивается асептический менингит.
Семейная средиземноморская лихорадка. Диагностика.
Диагноз ставится на основании клинической картины болезни и преимущественного заболевания людей определенных национальностей. При этом у детей и подростков она возникает довольно рано. Аналогичные заболевания происходят и у близких родственников. Кроме того, семейная средиземноморская лихорадка утихает на фоне беременности, частота проявлений и их интенсивность значительно снижаются.
В целом диагностика основывается на 5 пунктах.
Клиническая картина. Приступы лихорадки с болевым синдромом, неэффективность антибиотиков и антипиретиков, хорошее самочувствие во внеприступный период.
Лабораторные данные. Лейкоцитоз с нейтрофиллезом, ускорение СОЭ, снижение активности миелопероксидазы нейтрофилов и повышение ее активности в крови в момент приступа; нормализация показателей вне приступа.
Генетическое исследование. Наиболее достоверный диагностический признак семейной средиземноморской лихорадки. Выявление гомозиготного носительства мутаций М680I, М694V, V726А делает диагноз периодической болезни 100%-ным. Однако здесь также возможны определенные трудности, когда при типичной клинике и анамнезе выявляется гетерозиготное носительство мутаций. Подобная ситуация может иметь место, когда в одной из аллелей гена пирина выявляется одна из вышеуказанных мутаций, а в другой — более редкая, не определяемая при стандартном типировании.
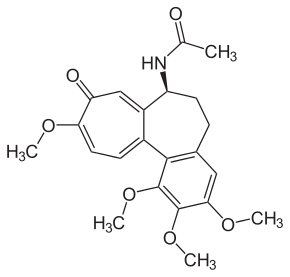
Эффект от терапии колхицином. Пробная терапия колхицином в качестве диагностического критерия необходима, когда нет возможности провести генетическое исследование или когда его результаты не дают полного подтверждения диагноза семейной средиземноморской лихорадки (гетерозиготные носители мутаций М680I, М694V, V726А или носители более редких мутаций). Наличие эффекта от терапии подтверждает диагноз.
Семейная средиземноморская лихорадка. Лечение.
Семейная средиземноморская лихорадка. Осложнения.
Семейная средиземноморская лихорадка — моногенное, рецессивно наследуемое, аутовоспалительное заболевание, характеризующееся возвратными приступами лихорадки и асептического полисерозита (перитонит, плеврит, синовит).
Основными осложнениями семейной средиземноморской лихорадки являются:
— системный амилоидоз, наиболее часто — амилоидоз почек, с развитием прогрессирующей хронической почечной недостаточности.
— спаечная болезнь, с развитием спаечной кишечной непроходимости, а также вторичного бесплодия.
— отставание физического и полового развития.
К развитию указанных тяжелых осложнений и инвалидизации приводит отсутствие лечения. Регулярное диспансерное наблюдение, совместно с непрерывным лечением, практически устраняют опасность развития у больных осложнений.
Амилоидоз.
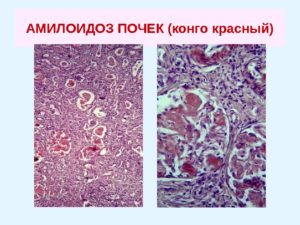
Каждый приступ семейной средиземноморской лихорадки сопровождается образованием воспалительных белков. Белки в больших количествах попадают в кровь, и задача организма от них кровь очистить. Каждый последующий приступ делает эту задачу сложнее. Образование нерастворимого белка (амилоида) — неизбежный побочный продукт очистки. Образование и отложение в тканях амилоида приводит к развитию амилоидоза.
Основными симптомами амилоидоза почек являются:
— наличие белка в моче;
— артериальная гипертония;
— отеки;
— хроническая почечная недостаточность.
Семейная средиземноморская лихорадка. Где лечиться в России.

В последние годы в России увеличивается частота обращений пациентов с характерными симптомами, скорее всего, это связано с усилением миграции населения и увеличением числа межэтнических браков. В учетом большой диаспоры армян в РФ, сопоставимой с населением Армении, можно ожидать, что в России приблизительно такое же количество больных семейной средиземноморской лихорадкой, как в Армении (около 17000 человек). .
В России на базе Научного центра Здоровья Детей г. Москва совместно с профильными лечебными учреждениями Армении разрабатывается программа, направленная на своевременное выявление и лечение детей с семейной средиземноморской лихорадкой.
Рассказ о болезни и жизни с ней пациентки с семейной средиземноморской лихорадкой.
Мы живем в XXI веке. В наши дни даже рак — не приговор. Что уж говорить о лихорадке, каковой на самом деле и является периодика? Вам предстоит трудная борьба за свое здоровье — и за свою жизнь. Но упорство даст результат, главное не сдавайтесь. Ни секунды не сдавайтесь. И вы не только забудете о своей болезни как о страшном сне, но и защитите свое потомство от нее.
Если кто-то из ваших родственников в прошлом погиб от этого недуга, не примеряйте печальный опыт на себя. Развитие в лечении этой болезни за последние десять лет сильно прогрессировало.
Что нужно знать родным больного:
1) Обычно вы узнаете, что больному плохо, потому что он кричит от боли. Но на это не всегда есть силы. Когда приступ достигает пика, слабеют речевые центры. ИНОГДА ОН НЕ МОЖЕТ ПОЗВАТЬ ВАС НА ПОМОЩЬ, ХОТЯ СПОСОБНОСТЬ ГОВОРИТЬ НЕ УТРАЧЕНА ПОЛНОСТЬЮ. Так что, если больной прилег, поинтересуйтесь самочувствием. Если он не кричит, это не значит, что ему не больно. Иногда боль не острая, а тяжелая, тупая. Она разлита по всему телу сразу. Да, да, болит ВЕЗДЕ и СРАЗУ, и на вопрос что с тобой, внятного ответа вы можете не услышать.
2) Характерные признаки, что начался приступ: больной подавлен, заторможен, иногда агрессивен, насколько позволяют силы. ЦВЕТ ЛИЦА сильно потемнел. Синева под глазами усилена. Больной как бы полупарализован. Потрогайте руки и ноги — они ледяные. Приставать с расспросами не всегда результативно — примите во внимание, что из-за сильной боли сознание больного в полутумане.
3) Что делать: срочно согревать. Шерстяные носки (даже в жару). Шаль под ребра — область солнечного сплетения — наша цель. Грелку в руки. Горячее питье. ВНИМАНИЕ: при жалобе на тошноту ни в коем случае не давать противорвотное. Напротив, нужно немедленно вызвать рвоту. За желудок не беспокойтесь — он тут не причем. Сильные сокращения желудка позволяет снять спазм зоны солнечного сплетения, она как бы каменеет во время приступа, и потому рвота — это серьезное облегчение для больного.
Часто родственники пытаются накормить больного обезболивающими. Отговаривать не буду — иногда это лучше, чем адские боли. Но просто запомните раз и навсегда: АНАЛЬГИН НЕ ПОМОГАЕТ. Он лишь немного облегчает боль, и то ненадолго. Сильные обезболивающие возравщают больного к жизни на время приступа, но со временем убивают нервную систему. Лучше всего большая доза аскофена или цитрамона, но не одна таблеточка, от которой толку как от пареной репы, а по две таблетки по мере необходимости в течение дня. ВНИМАНИЕ: при сильном приступе аскофен тоже не помогает, это актуально лишь при легкой форме, в самом начале развития приступа, когда больной в состоянии сам подняться, сесть-встать.
4) Не давите на больного. Приказной тон, ворчливость, придирки — самое худшее из всего, что вы можете ему дать, даже если вы уверены, что ваш резкий тон поможет ему собраться. Не поможет. Периодика нужно расслаблять и давать ему чувство безопасности, а не напряжения.
5) У вашего больного, скорее всего, ужасный характер. Но если вы будете подавлять его, он станет еще хуже. Панацея при таком недуге — выражение страхов и утешение, а не призывы к тому, чтобы больной собрался. Поймите, если человек всю жизнь терпит такие страдания, он уже на многое способен и обладает силой воли. Но для выздоровления он очень нуждается в расслаблении нервной системы. Научитесь быть тактичным и все свои претензии предъявляйте настойчиво, но без агрессии.
6) После того, как приступ прошел, человек полностью восстанавливается. Иногда это может случиться в течение одного дня. И даже нескольких часов. Больной вдруг преображается и резко приходит в себя. Эта внезапность исцеления часто вводит в заблуждение родных — они думают, что больной симулирует недуг. Это не так.
7) Запомните раз и навсегда, мы живем в ХХI веке. Это раньше периодику диагностировали тогда, когда больному уже отказывали почки. Современные методы диагностики и результаты изучения болезни позволяют поставить правильный диагноз на стадии, когда она еще не опаснее бронхита — даже без анализов (то есть в целом ничего хорошего, но полное исцеление реально). Так что прекратите паниковать, берите себя в руки и помогите родственнику лечиться.
Если болеете вы сами:
1) Научитесь определять начало приступа. За день-два вы становитесь раздражительны, вас тревожат мрачные мысли, плохое настроение. Будьте внимательны к цвету лица и температуре конечностей. Если успеете купировать приступ на этой стадии, в этот раз все может обойтись.
Глоток-два коньяка (ни в коем случае не больше — станет хуже), приятное занятие, покой, и желательно сон. Если все это сделать вовремя, возможно, в этот раз вам не придется рыдать от неспособности самостоятельно дойти до ванной. Дамы! будьте внимательны к своему циклу!
2) Всякое мрачное настроение нужно немедленно остановить. Боритесь с подавленным состоянием, как можете. Но не вгоняйте его в себя, а выкидывайте. Рисование, танцы, иногда даже скандалы — не держите в себе агрессию, раздражительность. От битья посуды лучше отказаться))) Найдите мирный способ. Научитесь петь — смех смехом, а это помогает расслабить солнечное сплетение, что очень важно. Обратитесь к профессионалу, к психотерапевту. Фобии, подавленность, скрытая агрессия — вот вам официальный повод для обращения. По мере того, как вы избавитесь от иррациональных страхов, болезнь начнет таять и очень скоро вас покинет навсегда.
3) Если приступ уже начался — берите себя в руки и помогите себе сами. Попросите родных принести горячее питье — не крошечную чашечку, а огромную кружку чая. Включите хороший, добрый фильм. Повяжите под ребрами шерстяную шаль — поверьте, это сильно облегчит ваше состояние. А если успеть все сделать вовремя — вы успеете остановить приступ. Если не успели и приступ разыгрался, вам не хочется жить и все такое — помните, вы не умираете. Это пройдет. Пройдет бесследно через несколько часов. Ждите этого момента, и вам станет легче.
4) Не злитесь на родственников. Они правда иногда не понимают, почему вы три дня валяетесь на кровати, особенно, если вы не катаетесь по полу от боли. И поймите, что им тоже нелегко. Они хотят видеть вас бодрым, добрым и успешным человеком. А вы вдруг становитесь вялым, раздражительным и непонятно почему вдруг не в состоянии работать столько, сколько работаете в обычное время. И они часто считают вас симулянтом: не могут понять, как это вы пролежав три в постели вдруг встали и как ни в чем не бывало побежали по делам. Ситуация ужасная, но не безнадежная. Главное помните — это не враги. Это ваши родственники.
5) Чем скорее вы научитесь избегать состояния подавленности, тем быстрее забудете о боли под ребрами в частности и о периодике вообще. Первое время даже можно побыть агрессивным, но помните, что в идеале вам необходимо научиться:
а) выражать свои чувства в не оскорбительной для родных форме, но твердо и несгибаемо.
б) не обижаться — чем скорее научитесь, тем целее будете.
в) не испытывать чувства вины за свои чувства и не позволять родным играть на нем.
г) не испытывать чувства вины за то, что имеете право быть собой, а не тем, что из вас лепит ваша любящая семья. Тем более, что вы скорее всего не желаете заниматься каннибализмом или торговать запрещенными препаратами, а хотите всего лишь одеваться не так, как принято в вашей семье. Мелочь, а очень важно для вашего выздоровления: быть собой. Не думайте, что это несерьезная рекомендация при серьезном заболевании.
5. Колхицин. Колхицин стал панацеей для многих. Со временем он останавливает приступы, и это прекрасно. Но он не лечит депрессивные состояния и фобии, которые лежат в основе расстройства, и они остаются миной замедленного действия, которая может рвануть в любой момент. Не смотря на пять лет приема колхицина. Вывод — колхицин колхицином, а работать нужно с первопричиной. То есть с фобиями и скрытой агрессией.
7 Смена климата. Спросите любого в Армении и он вам скажет, что смена климата лечит болезнь. Спросите любого знающего армянина, и он вам скажет что смена климата помогает на полгода. В чем истина? В том что помогает не сам переезд в другую климатическую зону. Помогает смена обстановки. И приступов не будут ровно столько времени, пока больного не будут подавлять как личность. Как только он попадет в ситуацию прессинга (кстати, столь популярную в добропорядочных армянских семьях), и приступов не избежать.
9 Кто в группе риска. Думаете, армяне-евреи-арабы? хе-хе-хе… В группе риска все, чьи бабушки и дедушки пережили гуманитарную катастрофу, конкретнее, ситуацию при которой гибло большое количество народа сразу. В особой группе риска — представители семей, в которых не принято обсуждать пережитое горе, а говорится только обо всем светлом и хорошем. Замалчивание коллективного стресса не приводит ни к чему хорошему.
10 О том, с чего мы начали. Периодика не приговор. Все в ваших руках.
Периодическая болезнь (синонимы: семейная средиземноморская лихорадка, доброкачественный пароксизмальный перитонит, рецидивирующий полисерозит, еврейская болезнь, армянская болезнь) — наследственное аутосомно-рецессивное заболевание, распространенное сред
Периодическая болезнь (синонимы: семейная средиземноморская лихорадка, доброкачественный пароксизмальный перитонит, рецидивирующий полисерозит, еврейская болезнь, армянская болезнь) — наследственное аутосомно-рецессивное заболевание, распространенное среди представителей древних народов Средиземноморья. Наиболее часто периодическая болезнь (ПБ) встречается у евреев-сефардов, армян, арабов, греков, турков, народов Кавказа и т. д., отсюда другие названия заболевания. Встречаемость ПБ среди евреев-сефардов по разным данным составляет от 1:250 до 1:2000 (частота носительства мутантного гена от 1:16 до 1:8), среди армян — от 1:100 до 1:1000 (частота носительства — от 1:7 до 1:4).
Среди 15 детей с ПБ, наблюдавшихся в Российской детской клинической больнице (РДКБ) в течение последних лет, 8 были армянами, 4 — дагестанцами, 1 — грек, 1 — с чеченскими и еврейскими корнями, 1 — русский.
Этиология и патогенез
В основе ПБ лежит точечная мутация в гене белка пирина, расположенного в коротком плече 16-й хромосомы (16q) рядом с генами аутосомно-доминантного поликистоза почек и туберозного склероза. Пирин — белок первичных гранул нейтрофилов, активно участвующий в регуляции процессов воспаления. Считается, что пирин стимулирует выработку противовоспалительных медиаторов, позволяет контролировать хемотаксис, стабилизирует мембрану гранулоцитов. Нарушение структуры этого белка, имеющее место при ПБ, приводит к повышению выработки провоспалительных медиаторов в лейкоцитах, активации микротубулярного аппарата и спонтанной дегрануляции первичных гранул лейкоцитов, активации молекул адгезии и усиленному хемотаксису лейкоцитов, результатом чего является воспаление.
ПБ протекает в виде приступов, основой которых является спонтанная или спровоцированная дегрануляция нейтрофилов с выбросом медиаторов и развитием асептического воспаления преимущественно на серозных и синовиальных оболочках. В периферической крови повышается количество нейтрофилов и острофазовых белков (СРБ — С-реактивный реактивный белок, SAA — сывороточный белок амилоида А и др.). Раздражение медиаторами воспаления рецепторов приводит к развитию болевого синдрома, а воздействие большого количества эндогенных пирогенов на центр терморегуляции — к развитию лихорадки.
Клиническая картина и течение
Клинически ПБ проявляется возникающими через определенные интервалы (дни — недели — месяцы) стереотипными приступами лихорадки. Лихорадке могут сопутствовать болевые синдромы, связанные с развитием неспецифического воспаления в серозных и синовиальных покровах. В зависимости от пенетрантности генов эти синдромы могут быть изолированными или сочетаться, но каждый из них сохраняет свой ритм. Любая атака сопровождается лейкоцитозом, увеличением СОЭ и других воспалительных белков, повышением a- и b-фракции глобулинов, снижением активности миелопероксидазы нейтрофилов. Вне приступа дети чувствуют себя хорошо, лабораторные показатели постепенно нормализуются.
Длительность лихорадочного и абдоминального вариантов ПБ обычно составляет от 1 до 3 дней, реже удлиняется до 1–2 нед.
Перитонит, как и суставной синдром, наиболее закономерен для детского возраста.
Торакальный вариант с плевральным синдромом встречается реже — около 40% случаев, изолированно — в 8%, в сочетании с абдоминальным синдромом — в 30%. При торакальном варианте развивается одно-двусторонний плеврит со стерильным выпотом. Длительность этого синдрома — 3–7 дней. Как правило, таким больным ошибочно устанавливается диагноз плеврита или плевропневмонии.
Кожные изменения во время приступа ПБ встречаются в 20–30% случаев. Наиболее типичной является рожеподобная сыпь, но могут встречаться пурпурные высыпания, везикулы, узелки, ангионевротические отеки. Иногда клинически ПБ протекает подобно аллергической реакции вплоть до отека Квинке и крапивницы.
Другими проявлениями ПБ могут быть головная боль, асептический менингит, перикардит, миалгия, гепатолиенальный синдром, острый орхит.
Манифестация заболевания может приходиться на различный возраст. Описаны случаи довольно позднего манифестирования ПБ, после 20–25 лет. По нашим наблюдениям, у большинства больных первый приступ ПБ отмечался в возрасте 2–3 лет (9 пациентов), у 1 — с рождения, у 2 — в 0,5–1,5 года, у 2 — в 4–5 лет, у 1 — в 11–12 лет.
Частота и периодичность приступов варьируются у разных больных в широких пределах: от нескольких раз в неделю до 1–2 раз в несколько лет. У большинства больных приступы имеют довольно стабильный ритм. Однако в литературе описаны случаи, когда приступы могли прекратиться на несколько лет или, наоборот, возобновиться после длительного перерыва под воздействием внешних факторов (смена места жительства, женитьба или замужество, рождение ребенка, служба в армии и др.). У наших пациентов периодичность приступов была довольно постоянной: у 1-го — 2 раза в нед, у 4-х — 1 раз в нед, у 5-ти — 1 раз в 2–3 нед, у 2-х — 1 раз в мес, у 1 — 1 раз в 2–3 мес, у 1-го — 1 раз в 6–12 мес.
Через некоторое время от начала манифестации у большинства больных отмечается гепатомегалия, которая, по нашим наблюдениям, может варьироваться от +1 до +5 см. Постепенно развивается и спленомегалия, величина которой у некоторых пациентов достигала +7 см. Однако увеличение печени и селезенки выявляется не у всех больных. Очевидно, эти процессы зависят от частоты и количества перенесенных приступов и развития амилоидоза.
Амилоидоз как осложнение периодической болезни
Амилоидоз (от лат. amylum — крахмал) — собирательное понятие, включающее группу заболеваний, характеризующихся внеклеточным отложением белков в виде характерных амилоидных фибрилл. Эти нерастворимые фибриллярные белки могут быть локализованы в одном специфическом месте или могут быть распространены в различных органах, в том числе таких жизненно важных, как почки, печень, сердце и др. Такое накопление приводит к органной дисфункции, недостаточности органа и в конечном итоге смерти.
Структура амилоида идентична при всех его типах и представляет собой жесткие неразветвляющиеся фибриллы диаметром около 10 нм, обладающие складчатой β-кросс-конформацией, благодаря которой возникает эффект двойного лучепреломления в поляризованном свете при окраске Конго-красным. Окраска щелочным Конго-красным является наиболее распространенным и доступным методом выявления амилоида.
Амилоид состоит из фибриллярных белков (фибриллярный компонент, F-компонент) и гликопротеидов плазмы крови (плазменный компонент, P-компонент). Предшественники F-компонента различаются при различных видах амилоидоза (на сегодняшний день известно до 30 белков-предшественников, они определяют тип амилоидоза); предшественник Р-компонента один — сывороточный амилоидный Р-компонент (SAP), схожий с α-глобулином и СРБ.
Фибриллы амилоида и плазменные гликопротеиды образуют комплексные соединения с хондроитинсульфатами ткани с участием гематогенных добавок, среди которых основными являются фибрин и иммунные комплексы. Связи между белковыми и полисахаридными составляющими в амилоидном веществе особо прочные, что объясняет отсутствие эффекта при воздействии на амилоид различных ферментов организма, т. е. амилоид нерастворим.
При ПБ основой формирования фибриллярного компонента амилоида является сывороточный острофазовый белок SAA. SAA является a-глобулином, близким по своим функциональным свойствам к СРБ. SAA синтезируется клетками разных типов (нейтрофилами, фибробластами, гепатоцитами), его количество повышается во много раз при воспалительных процессах и опухолях. У человека выделено несколько типов SAA, и только фрагменты некоторых из них входят в состав амилоидных фибрилл, что, возможно, объясняет развитие амилоидоза только у части больных, несмотря на повышенную выработку SAA. Из сывороточного SAA-предшественника в тканях образуется АА-белок (белок амилоида А), который и является основой амилоидных фибрилл. Поэтому тип амилоидоза, развивающегося при ПБ, называется АА-амилоидоз.
Таким образом, основой развития амилоидоза при ПБ является избыточное образование белка-предшественника SAA. Но для образования амилоидного белка необходимы клетки, которые будут его синтезировать — амилоидобласты. Эту функцию выполняют в основном макрофаги-моноциты, а также плазматические клетки, фибробласты, ретикулоциты и эндотелиальные клетки. Макрофаги перерабатывают АА-белок в полноценные амилоидные фибриллы на своей поверхности и откладывают его в межуточной ткани. Поэтому наибольшее накопление амилоида при ПБ отмечается в органах, где макрофаги занимают фиксированное положение: почки, печень, селезенка. Постепенно все увеличивающиеся отложения амилоида приводят к сдавливанию и атрофии паренхиматозных клеток, склерозу и недостаточности органа.
Амилоидоз при ПБ болезни развивается по различным данным у 10–40% больных. Некоторые пациенты, несмотря на довольно частые приступы, не развивают амилоидоз вовсе. Вероятно, развитие амилоидоза зависит от особенностей строения белка-предшественника у данного пациента и генетической способности макрофагов синтезировать амилоид.
Несмотря на то, что амилоидоз может развиваться в любом органе и ткани, амилоидное поражение почек играет определяющую роль для прогноза и жизни больного ПБ. При развитии АА-амилоидоза почки поражаются в 100% случаев.
В почках роль амилоидобластов выполняют мезангиальные и эндотелиальные клетки.
В процессе отложения амилоида в почечной ткани и вызванного им поражения органа можно проследить определенную стадийность. Выделяют 4 стадии амилоидоза почек: латентную (диспротеинемическую), протеинурическую, нефротическую (отечную) и уремическую (азотемическую).
В латентную стадию изменения в почках незначительны. Отмечаются нарушения гломерулярного фильтра в виде очагового утолщения, двухконтурности мембраны и аневризм ряда капилляров. В гломерулах амилоида нет или он обнаруживается не более, чем в 25% клубочков.
Ведущим в патогенезе этой стадии амилоидоза является значительный синтез и повышение в плазме крови концентрации белков-предшественников амилоидоза, т. е. диспротеинемия. Клинически у детей на фоне приступов ПБ может развиваться гипохромная железодефицитная анемия, гиперпротеинемия, диспротеинемия с увеличением глобулинов α2, β и γ, отмечается высокое содержание фибриногена и сиалопротеинов. Характерны увеличение и уплотнение печени и селезенки.
Изменения в моче поначалу отсутствуют или носят транзиторный характер, однако со временем протеинурия становится постоянной и более выраженной, часто наблюдается микрогематурия и цилиндрурия. Появление постоянной протеинурии характеризует переход во вторую, протеинурическую, стадию.
В протеинурической стадии амилоид появляется не только в пирамидах, но и в половине клубочков почек в виде небольших отложений в мезангии, отдельных капиллярных петлях, а также артериолах. Отмечается выраженный склероз и амилоидоз стромы, сосудов, пирамид и интермедиарной зоны, что приводит к атрофии многих глубокорасположенных нефронов.
Продолжительность этой стадии, как и предыдущей, колеблется от нескольких месяцев до многих лет. По мере нарастания тяжести амилоидоза усугубляются лабораторные показатели выраженной активности процесса: значительная протеинурия и диспротеинемия, гиперфибриногенемия, СРБ, гиперкоагуляция. Дальнейшее отложение амилоида в почечной ткани и нарастающая протеинурия приводят к развитию отечного синдрома, появление которого свидетельствует о переходе заболевания в третью, отечную, стадию.
В отечную (нефротическую) стадию амилоидоза количество амилоида в почках увеличивается. Пораженными оказываются более чем 75% гломерул. Прогрессирует склероз интерстиция и сосудов, в пирамидах и интрамедиарной зоне склероз и амилоидоз имеют выраженный диффузный характер.
Клинически эта стадия амилоидоза представлена полным нефротическим синдромом, хотя иногда может наблюдаться неполный (безотечный) нефротический синдром. Протеинурия становится массивной и, как правило, неселективной; нарастают циллиндры. Гематурия бывает редко и, как правило, незначительна. Нарастают гепатоспленомегалия, гипопротеинемия, усиливаются диспротеинемия с дальнейшим повышением уровня α1-, α2-, и γ-глобулинов, гиперфибриногенемия, гиперлипемия. Со временем появляется артериальная гипертензия, нарастает азотемия, прогрессирует почечная недостаточность.
Уремическая (азотемическая) стадия развивается в финале заболевания. В связи с нарастающим амилоидозом и склерозом наблюдаются гибель большинства нефронов, их замещение соединительной тканью, развивается ХПН (хроническая почечная недостаточность).
Клиническими особенностями ХПН при амилоидозе, отличающими ее от ХПН вследствие других заболеваний, является сохранение нефротического синдрома с массивной протеинурией, часто определяются большие размеры почек, характерно развитие гипотензии.
Часто выражен ДВС-синдром (синдром диссеминированного внутрисосудистого свертывания крови) в виде пурпуры, носовых, желудочных и кишечных кровотечений. Возможны тромбозы почечных сосудов с развитием инфарктов ишемического или геморрагического типа.
Мы наблюдали развитие амилоидоза у 4-х детей с ПБ (26% наблюдаемых больных). Транзиторная протеинурия у них появлялась на 7–8 год от манифестации заболевания, через 2–3 года она принимала постоянный характер. У 2-х детей через 1,5–2 года после установления постоянной протеинурии развился нефротический синдром, который у одного ребенка перерос в ХПН.
Развитие амилоидоза в определенной степени зависит от количества перенесенных ребенком приступов ПБ. Среди наших пациентов амилоидоз почек выявлялся у тех, кто перенес более 130–150 приступов, тогда как у детей с меньшим количеством приступов признаков амилоидоза и поражения почек не отмечалось. Причем дети с нефротическим синдромом перенесли наибольшее число приступов — около 240 и 260. Следует отметить, что подобная закономерность не является абсолютной и амилоидоз может развиться и при меньшем количестве приступов ПБ.
Диагностика периодической болезни и амилоидоза
При типичном течении периодической болезни ее диагностика не представляет трудностей. Наибольшая проблема заключается в незнании большинством врачей этой патологии, что приводит к плохой выявляемости даже при наличии симптомов.
Диагностика ПБ основывается на 5 пунктах.
При постановке диагноза ПБ в большинстве случаев у врача возникает настороженность в отношении амилоидоза. Но часто первые подозрения на АА-амилоидоз могут возникнуть у педиатра при лечении больных с нефротическим синдромом, резистентных к стандартной глюкокортикоидной терапии.
Только изучение материалов биопсии с обязательным окрашиванием Конго-красным и поляризационной микроскопией позволяет поставить окончательный диагноз АА-амилоидоза. Помимо этого для диагностики можно использовать специфические антитела к АА-фибриллам. Наиболее достоверной является биопсия почки. Частота выявления АА-амилоидоза в этом случае достигает 90–100%. Чем более распространен процесс, тем больше вероятность выявления АА-амилоида в других местах (желудочно-кишечный тракт (ЖКТ) — слизистая и подслизистая, слизистая десны, прямая кишка, жировая биопсия). Наиболее информативной среди непочечных биопсий является биопсия стенки ЖКТ и прямой кишки, при которой вероятность выявления амилоида составляет 50–70%.
Лечение
При периодической болезни основой терапии является назначение колхицина. Колхицин обладает антимитотическим эффектом в отношении амилоидобластов при периодической болезни — макрофагов и стабилизирует мембрану нейтрофилов, препятствуя выбросу пирина. Колхицин назначается пожизненно в дозе 1–2 мг/сут. Он хорошо переносится, иногда возникают диспептические явления, которые не требуют полной отмены препарата. Колхицин в большинстве случаев полностью предотвращает появление приступов ПБ или значительно снижает их частоту и выраженность, предотвращает развитие амилоидоза почек, снижает выраженность его проявлений. При почечной недостаточности дозу снижают исходя из степени снижения клубочковой фильтрации. Препарат может быть временно отменен при острых инфекциях у ребенка.
Нами наблюдался мальчик, который был направлен в РДКБ с генетически установленным диагнозом периодической болезни в возрасте 16 лет. Приступы ПБ отмечались у него с 4-летнего возраста, протекали с периодичностью 1 раз в 2–3 нед в виде лихорадки с абдоминальным болевым синдромом, 1–2-кратной рвотой, головной болью и выраженной слабостью. Приступы длились около суток, затем в течение 1–2 дней была столь выраженная слабость, что мальчик не мог встать с постели, не посещал школу. Признаков амилоидоза не выявлялось.
В РДКБ ребенку был назначен колхицин в дозе 2 мг/сут. За последующие 2 года наблюдения число приступов резко снизилось до 1–2 раз в год, а в течение последних 10 месяцев не было ни одного. Сейчас юноша успешно обучается в вузе, живет в общежитии в другом городе, чувствует себя хорошо.
В лечении ПБ и профилактике амилоидоза необходима организация правильного питания ребенка. Увеличение общего количества белка в рационе стимулирует амилоидогенез, тогда как белок печени и мышцы сердца ингибируют его. Рекомендуется диета со сниженным на 50% содержанием животного (особенно казеина) и растительного белков и увеличением продуктов, содержащих крахмал. Диета должна быть достаточно обогащенной фруктами, овощами и другими шлакогонными продуктами. Белок предпочтительнее давать ежедневно (100 г печени, сырой или кулинарно обработанной). Печень употребляют годами, в виде повторных многомесячных курсов. Используют гепатотропные препараты повторными курсами: по 2–4 мес Эссенциале, Липоевую кислоту.
Прогноз
При своевременной постановке диагноза и назначении колхицина прогноз ПБ благоприятен.
При отсутствии терапии наибольшую опасность представляет развитие почечного амилоидоза, который, по сути, является единственной причиной смерти больных с ПБ. Анализ заболеваемости у взрослых и детей показывает, что при естественном течении периодической болезни приблизительно у 50% больных терминальная стадия почечной недостаточности развивается через 5 лет от момента появления протеинурии, у 75% — в течение 10 лет.
По вопросам литературы обращайтесь в редакцию.
А. В. Малкоч, кандидат медицинских наук
РГМУ, Москва
Читайте также:
