
Болезнь крейтцфельдта-якоба на мрт
а) Терминология:
1. Сокращения:
• Болезнь Крейтцфельдта-Якоба (БКЯ)
• Спорадическая болезнь Крейтцфельдта-Якоба (сБКЯ)
• Вариантная болезнь Крейтцфельдта-Якоба (вБКЯ)
2. Определение:
• Быстро прогрессирующее, смертельное нейродегенеративное заболевание, вызываемое прионами (белковые инфекционные частицы, не содержащие ДНК и РНК):
о Трансмиссивная губчатая энцефалопатия
1. Общие характеристики болезни Крейтцфельдта-Якоба (БКЯ):
• Лучший диагностический критерий:
о Повышение интенсивности сигнала от базальных ганглиев (БГ), таламусов и коры больших полушарий на Т2-ВИ прогрессирующего характера
• Локализация:
о Преимущественное поражение серого вещества (СВ)
- БГ: хвостатые ядра и скорлупа > бледные шары (БШ)
- Таламусы (часто при вБКЯ)
- Кора больших полушарий (наиболее часто в процесс вовлекаются лобные, теменные и височные доли):
Вовлечение коры часто имеет асимметричный характер
Вариант Хайденхайна: затылочные доли
Вариант Браунэлл-Оппенгеймера: мозжечок
о Белое вещество (БВ) обычно не поражается

2. КТ признаки болезни Крейтцфельдта-Якоба (БКЯ):
• Бесконтрастная КТ: обычно нормальная картина:
о При КТ обследованиях в динамике возможно выявление быстропрогрессирующей атрофии и расширения желудочков
4. Радионуклидная диагностика болезни Крейтцфельдта-Якоба (БКЯ):
• ПЭТ: регионарный гипометаболизм глюкозы коррелирует с участками поражения на патоморфологических препаратах
• ОФЭКТ с N-изопропил-n-(I-123)-иодамфетамином
о ↓ поглощения РФП и ↓ абсолютных значений rCBF в различных частях коры больших полушарий
5. Рекомендации по визуализации:
• Лучший инструмент визуализации: МРТ с ДВИ и FLAIR
в) Дифференциальная диагностика болезни Крейтцфельдта-Якоба (БКЯ):
1. Гипоксически-ишемическое поражение:
• Наблюдается вовлечение в процесс БГ и парасагиттальных областей коры
• Гиперинтенсивные очаги поражений в БГ на Т1-ВИ и Т2-ВИ
• ДВИ + симметричное вовлечение в процесс СВ
2. Синдром осмотической демиелинизации:
• Экстрапонтинная локализация: повышение интенсивности сигнала от скорлупы и хвостатых ядер на Т2-ВИ
• В острый период - ограничение диффузии на ДВИ
3. Синдром Лея:
• В первую очередь характерен для детского возраста
• Повышение интенсивности сигнала от скорлупы и БШ на Т2-ВИ
4. Другие причины деменции:
• Болезнь Альцгеймера
• Деменция при болезни двигательного нейрона
• Лобно-височная деменция
• Мультиинфарктная деменция
5. Кортикобазальная дегенерация:
• Потеря нейронов в черной субстанции, коре лобно-теменных отделов больших полушарий и полосатых телах (атрофия БГ может быть выражена незначительно)
• МРТ: симметричная/асимметричная атрофия пре- и постцентральных извилин; выраженное поражение парасагиттальных отделов
• Субкортикальный глиоз: повышение интенсивности сигнала на Т2-ВИ
6. Болезнь Вильсона:
• Поражения БВ и субкортикального СВ (БГ, зубчатые ядра, ствол мозга); вариабельный характер повышения интенсивности сигнала на Т2-ВИ
• Гипоинтенсивные поражения на Т1-ВИ (реже гиперинтенсивные)
7. Атеросклероз:
• Вовлечение в процесс БГ: обычно асимметричный и мультифокальный характер
• (а не диффузный, что характерно для БКЯ)
• Локальные гиперинтенсивные очаги в глубоком БВ
• Отсутствие ограничения диффузии на ДВИ, исключая острую фазу
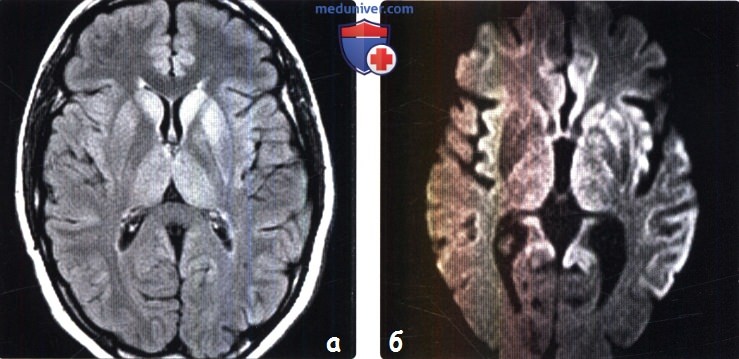
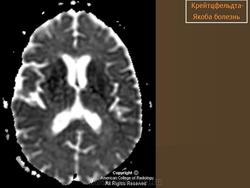
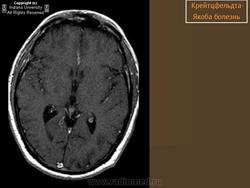
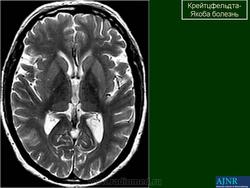
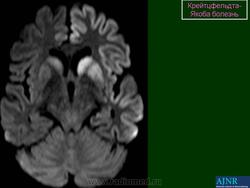
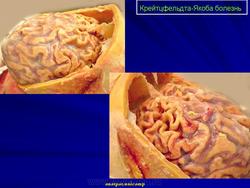
(а) MPT, FLAIR, аксиальный срез: определяется асимметричное повышение интенсивности сигнала от головок хвостатых ядер, больше слева, и левой скорлупы.
(б) МРТ, ДВИ, аксиальный срез: у того же пациента через несколько недель отмечается асимметричное повышение интенсивности сигнала от базальных ганглиев, больше слева, и от коры больших полушарий. При БКЯ более часто наблюдается асимметричное вовлечение в процесс коры, чем базальных ганглиев. У этого пожилого мужчины наблюдалась быстро прогрессирующая деменция и был поставлен диагноз вероятная БКЯ, так как при ЭЭГ были выявлены характерные признаки.
1. Общие характеристики болезни Крейтцфельдта-Якоба (БКЯ):
• Этиология:
о Прионные белки представляют собой неправильно свернутую изоформу (PrPSc) кодируемого в норме геномом хозяина белка (РгРс)
о PrPSc, введенный в здоровые клетки → запускает самовоспроизводящийся порочный цикл: РгРс → PrPSc
о сБКЯ: спонтанное образование РгРс → PrPSc или вследствие соматической мутации
о Семейная форма БКЯ (семБКЯ): мутации в гене PRNP О БКЯ ятрогенного характера: распространение инфекции из прионсодержащего материала:
- Хирургические инструменты, трансплантаты из твердой мозговой оболочки
- Трансплантация трупной роговицы, препараты гормона роста человека
о вБКЯ: губчатая энцефалопатия крупного рогатого скота передается людям через зараженную говядину:
- также известна как новый вариант БКЯ (нвБКЯ)
• Генетика:
о Может иметь наследственный, спорадический или приобретенный (инфекционный) характер
о 10-15% прионных заболеваний человека связаны с мутациями гена (PRNP) прионного белка (PrPc), локализованного на 20-й хромосоме и имеют доминантно-аутосомный тип наследования
• Ассоциированные аномалии
о ЭЭГ: периодические [высоковольтные] спайк-волна комплексы (ППВК) на фоне низковольтажной активности
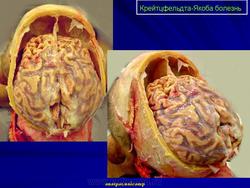
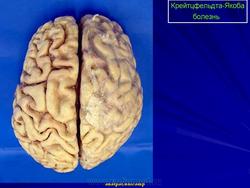
2. Макроскопические и хирургические особенности:
• Легкая атрофия коры больших полушарий
• Расширение желудочков
3. Микроскопия:
• Спонгиозная энцефалопатия: наиболее выражено поражение СВ:
о Выраженная потеря нейронов с признаками реактивного астроцитоза
о Заместительный глиоз
о Вакуолизация нейронов со спонгиозными изменениями
• У 10% пациентов с БКЯ наблюдается отложение амилоидных бляшек в мозжечке или полушариях большого мозга
• Вариабельный характер накопления PrPSc в ткани головного мозга
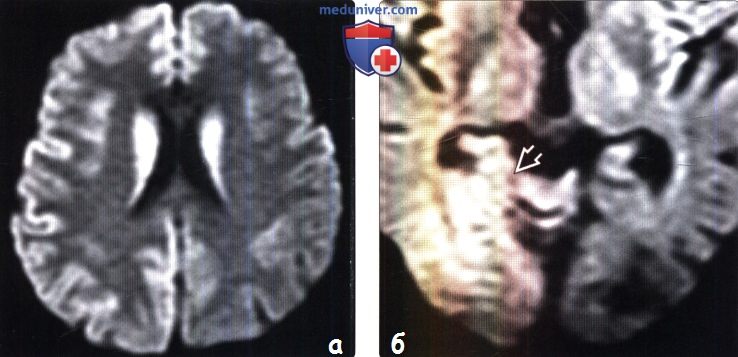
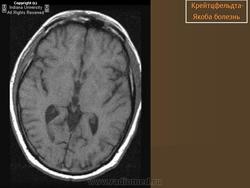
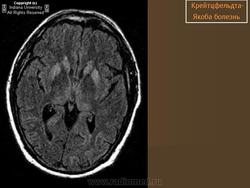
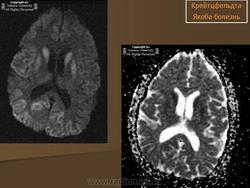
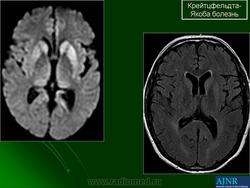
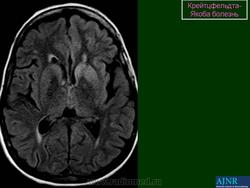
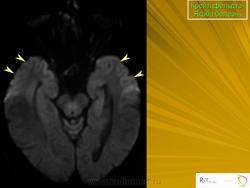
(а) МРТ, ДВИ, аксиальный срез: у данного пациента с БКЯ определяется выраженное повышение интенсивности сигнала от хвостатых ядер и коры вследствие ограничения диффузии. ДВИ является наиболее чувствительным методом для диагностики БКЯ.
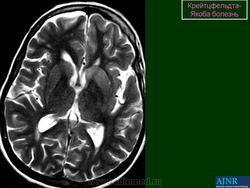
(б) МРТ, ДВИ, аксиальный срез: у пациента с начальными жалобами на зрительные нарушения в правой затылочной доле и левом островке визуализируются выражено гиперинтенсивные зоны. При варианте Хайденхайна БКЯ изначально возникают изолированные симптомы зрительных нарушений. При диагностической визуализации отмечается преимущественное вовлечение в процесс лобных долей.
д) Клиническая картина:
1. Проявления болезни Крейтцфельдта-Якоба (БКЯ):
• Наиболее частые признаки/симптомы:
о Прогрессирующая деменция в сочетании с миоклоническими судорогами и акинетическим мутизмом о Вариабельная картина нарушения пирамидных, экстрапирмидных и мозжечковых функций
• Клинический профиль:
о сБКЯ: нарушение функции мозжечка, быстро прогрессирующие когнитивные нарушения или их сочетание:
- 6 молекулярных подтипов: ММ1, ММ2, MV1, MV2, VV1 и VV2:
Варьируют в зависимости от возраста начала заболевания, продолжительности, ранних симптомов и патологических изменений
о вБКЯ: симптомы нарушения психических и чувствительных функций
- Вариант Хайденхайна БКЯ:
Изолированные зрительные признаки/симптомы (первоначально)
Преимущественная дегенерация затылочных долей
- Вариант Браунэлл-Оппенгеймера: мозжечковые признаки/симптомы
- Экстра пирамидный тип БКЯ:
Возможно обнаружение ↑ интенсивности сигнала от БГ
- Вовлечение структур пирамидной системы при прогрессировании заболевания
о Исследования СМЖ:
- Белки -биомаркеры СМЖ: белок 14-3-3, общий тау-белок (о-тау) и нейрон-специфическая энолаза (НСЭ)
- МРТ с ДВИ имеет более высокую точность, чем любые или все три биомаркера СМЖ
о Вибрационно-индуцированный конверсионный анализ СМЖ в режиме реального времени (RT-QUIC) для определения PrPsc:
- Большей чувствительностью обладает исследование обонятельного эпителия (соскоб со слизистой оболочки носа), по сравнению с СМЖ
2. Демография:
• Возраст:
о Молодой при вБКЯ, пожилой при сБКЯ (с 6-й по 7-й декады жизни)
• Этническая принадлежность:
о сБКЯ распространен повсеместно, среди всех рас
о Распространение вБКЯ территориально ограничивается Европой (все случаи отмечаются в Великобритании)
• Эпидемиология:
о В США частота встречаемости 1-1,5 на 1 млн
о сБКЯ (85%), семейные (15%), инфекционные/ятрогенные (менее чем 1 %)формы
3. Течение и прогноз:
• Длительный инкубационный период, но быстрое прогрессирование после появления клинических симптомов
• Быстро прогрессирующая деменция с наступлением летального исхода обычно в течение нескольких месяцев от начала заболевания:
о Медиана выживаемости с момента появления симптомов до смерти составляет 4,5 месяца
о У 90% пациентов продолжительность жизни составляет
Редактор: Искандер Милевски. Дата публикации: 4.5.2019
Крейтцфельдта - Якоба болезнь




О. Хабиб
O. Habib
Губчатые энцефалопатии представляют собой инфекционные дегенеративные заболевания. Некоторые семейные формы обусловлены генетически. В ткани мозга обнаруживают патологические белковые частицы - прионы (PrPres), которые представляют собой устойчивую патологическую (лишенную нуклеиновой кислоты) копию прион-протеина (PrP), присутствующего в здоровой нервной ткани всех млекопитающих. PrP - это мембранный сиалогликопротеин, состоящий из 253 аминокислот; он имеет двухмерную пространственную структуру и кодируется единственным геном на коротком плече хромосомы 20. Патологическая форма белка обнаруживается только в пораженной инфекционной губчатой энцефалопатией (ИГЭ) ткани мозга. PrPres и PrP имеют различные пространственные структуры и свойства: PrPres нерастворим в воде, нечувствителен к протеазам, имеет в структуре большее количество b -цепей и меньшее количество a -спиралей. Предполагают, что конверсия PrP в PrPres происходит во время транскрипции. Пока неясно, является ли PrPres одновременно патогенным и вирулентным (так как не обнаружена нуклеиновая кислота, а в эксперименте удалось получить PrPres из PrP в ацеллюлярной системе) или в PrPres включен некий вирус (т.е., имеется нуклеиновая кислота), который и обусловливает инфекционную передачу, а PrPres дает только токсический эффект. Существование частицы с независимым геномом наиболее удачно объясняет наличие нескольких разновидностей PrPres. Одна из гипотез предполагает существование пока не распознанной молекулы, которая, прикрывая белок и вирус, мешает "увидеть" нуклеиновую кислоту.
Генетическая предрасположенность. Гену, ответственному за синтез PrP, свойствен полиморфизм. 129-ый Кодон может кодировать либо метионин, либо валин. 50% населения гетерозиготны по кодону 129 (метионин/валин), 40% гомозиготны по метионину и 10% - по валину. А поскольку все больные ИГЭ - гомозиготы, то утрату полиморфизма можно считать фактором, предрасполагающим к возникновению заболевания.
Разновидности PrPres. У пораженных ИГЭ овец верифицируют до 20 разновидностей PrPres и соответственно наблюдают различные инкубационные периоды и характер поражения ЦНС. Для ИГЭ коров характерен только один вид PrPres. Для болезни Крейцфельдта - Якоба (БКЯ) известны 4 разновидности (по иммуноблоттингу) PrPres.
Разновидности БКЯ. БКЯ является одним из видов человеческих ИГЭ. Помимо БКЯ к этой группе заболеваний относятся болезнь Куру ("смешливая" болезнь, эндемична для Новой Гвинеи, характерна 100% летальность), болезнь Гертсмана - Штройслера - Шайнкера (Gertsmann Straussler Scheinker) и фатальная семейная бессонница. По клинической картине и способу передачи различают следующие разновидности: классическая/спорадическая, семейная/генетическая, ятрогенная и британская ИГЭ.
Классическая форма описана Крейтцфельдтом (1920 г.) и Якобом (1921 г.). Заболеваемость в мире составляет 0,5-1 случай на 1 млн человек в год; во Франции регистрируется 30-50 случаев в год; 90% всех случаев БКЯ приходится на классическую форму. Заболевание обычно дебютирует в 55-75 лет, за 70 лет со времени описания зарегистрировано 20 исключений- ювенильных случаев (16-40 лет), которые рассматриваются сейчас как британский вариант. Характерно постепенное прогрессирование симптомов поражения ЦНС, в 13% случаев заболевание "разворачивается" в течение нескольких дней.
Клиническая картина: начальные симптомы в 35% случаев - депрессия и расстройство ментальных функций, в 34% - неврологические симтомы с преимущественным поражением зрительных функций и мозжечка; в 21% - сочетанные. Симптомы быстро прогрессируют: развивается глубокая деменция, часто в сочетании с мутизмом. Атаксия, тремор и ригидность обусловливают локомоторные расстройства вплоть до иммобилизации. Наступает смешанная кортико-ретинальная слепота; на поздней стадии нередки пирамидные расстройства, миоклонии и эпилептические припадки. Смерть наступает до истечения года с момента появления первых симптомов, в 90% случаев - к 5-му месяцу. Параклинические данные не специфичны: на ЭЭГ- периодические судорожные разряды, ликвор в норме (PrPres пока не определяют), на компьютерной томограмме - атрофия коры полушарий и мозжечка. Патоморфологически определяют уменьшение количества кортикальных нейронов, вакуолизацию ткани мозга, который приобретает вид губки; глиальную, в основном астроцитарную, пролиферацию. Признаки воспаления отсутствуют. PrPres верифицируют с помощью иммуногистохимического анализа и иммуноблоттинга. С 1968 г. инокулируют экстракт ткани мозга восприимчивым животным (приматам, грызунам, кошкам). Диагноз ставится на основании клинических проявлений и подтверждается патоморфологическими данными и заражением лабораторных животных.
Эпидемиология. Несмотря на обилие данных, пока не удается объяснить происхождение классической формы. Некоторое время назад обсуждалось алиментарное заражение в связи с увеличением заболеваемости в Исландии. Дискуссия возобновилась после верификации ИГЭ коров в 1986 г. Для заболевших людей характерны повышенный травматизм и частые хирургические вмешательства в анамнезе. Не исключено, что представители некоторых профессий - пастухи, фермеры, медики - подвержены повышенному риску заболевания.
Семейная (генетическая) форма составляет 5-6 % всех случаев БКЯ, наследуется по аутосомно-доминантному механизму. Чаще всего это мутация - замена аспартатовой кислоты на аспарагин в 178-й позиции молекулы PrP. Заболевание дебютирует на 5-10 лет раньше, чем при классической форме, течение заболевания такое же. Существуют изолированные этнические группы, где заболеваемость БКЯ в 30-100 раз выше, чем для населения в целом, что обусловлено особенностями генотипа и специфичностью мутаций. Это - ливийские евреи (Средний Восток и Средиземноморье), словацкие и чилийские общины, в которых распространены внутрисемейные браки.
Ятрогенная БКЯ появилась в последние годы, случаев мало, но очевиден их драматизм в психологическом, этическом и медико-юридическом аспектах. По типу заражения различают две группы. Центральное заражение происходит во время операций, хирургических манипуляций, через трансплантаты и хирургический инструмент. Известны 24 случая заражения при пересадке взятой у погибших от БКЯ твердой мозговой оболочки, 1 случай - при пересадке роговицы, 2 случая - после стереотаксической операции. Инкубационный период после инокуляции - от 7 до 120 мес. Закономерен вывод, что "инфекционное начало" (вирус?) очень устойчиво к воздействию традиционных антисептиков, которым обрабатывают инструменты и трансплантаты. Следовательно, они должны быть надежно обеззаражены, а оптимальным выходом является использование одноразовых инструментов. Известные случаи периферического заражения связаны с введением СТГ. Из всех 50 зарегистрированных 70-80% приходятся на Францию. 4 случая в Австралии связаны с введением ГТГ гипофиза для индукции овуляции. Сегодня очевидно, что замена естественного СТГ на рекомбинантный ( продукт генной инженерии) полностью устранит риск. Латентный период составляет 18-28 лет. Клиническая картина больше похожа на симптоматику болезни Куру - неврологические симптомы преобладают над интеллектуальной деградацией. У большинства больных наблюдают утрату полиморфизма на 129-м кодоне и характерное "поведение" прийона при выполнении иммуноблоттинга. В эксперименте доказана возможность заражения животных кровью больных. Таким образом безопасность донорских тканей и органов должна быть обеспечена, а их использование - обосновано.
Британский вариант. С 1995 по 1997 г. в Великобритании зарегистрировано 14 случаев БКЯ у молодых людей (и еще один во Франции). Общими для всех случаев были: нормальный ген PrP, гомозиготность по кодону 129, отсутствие каких-либо факторов риска, пространственно-временное совпадение (инкубация в несколько лет), характерное "поведение" PrPres при проведении иммуноблоттинга, напоминающее таковое при ИГЭ кошек, зараженных алиментарно. Появилась гипотеза о возможности алиментарного заражения при употреблении мяса больных животных. Ситуация прояснится в ближайшие два года с завершением экспериментов на трансгенетических мышах. Не исключено обнаружение неизвестных ныне защитных факторов.
Beauvais P. La maladie de Creutzfeldt-Jakob. La plus important des maladies a prion. La Presse Medical 1997;26:3787-92.
Описание и причины патологии
Во время исследований анамнеза заразившихся пациентов было установлено, что заболевание передается через инфицирование во время оперативного вмешательства, пересадку биологических волокон от человека к человеку, переливание продуктов крови, неправильное применение гормональных препаратов. Если употреблять в пищу мясо зараженных домашних животных, также может произойти инфицирование.
Прион не относится к биологическому патогену или к вирусному штамму. Он представляет собой агрессивный белок, который в некотором количестве содержится в клеточном строении клеток головного мозга, но имеет несколько измененную структуру. Патогенное белковое соединение, проникая в тело человека, разносится по организму с током крови, не разрушается, а откладывается на нейронах. Здоровая белковая часть клетки, вступая во взаимодействие с агрессором, преобразуется в подобную структуру, становясь патогеном. С накоплением в клетке таких форм происходит образование внешней нейронной бляшки, которая постепенно умерщвляет клеточную структуру органа.
Первые феноменальные проявления в организме происходят спустя долгий инкубационный период. Это связано с естественной длительностью распространения аномального элемента с током крови до мозговых структур, встраиванием в клеточный состав и преобразованием нормальных белков в измененную форму. В зависимости от способа инфицирования, длительность инкубации варьируется от одного года до 12-13 лет. Так, если ткани мозга были заражены через медицинские инструменты во время операции, первые изменения в мозге проявляются примерно через 14-20 месяцев. Если прион попал в организм с трансплантированными волокнами, первичная симптоматика будет выявлена через пять – шесть лет, а при внутримышечном введении гормональных средств, выделенных из секреции крупного рогатого скота, инкубационный этап может затянуться до 12-13 лет. Еще реже имеют место случаи болезни генетического характера, приводящие к стимуляции выработки собственного трансформированного белка.
Явные признаки заболевания
Чаще всего развитие недуга носит затяжной, плавный характер, но отмечаются и пациенты, страдающие стремительной вариацией – острой формой, когда болезнь начинается с резких поведенческих изменений и поражения когнитивных функций. Треть заболевших жалуются на такие состояния, как:
- болевой синдром в области головы;
- частые предобморочные ощущения;
- раздражительность, подавленное настроение;
- рассеянность внимания;
- прерывистость ночного сна, бессонница;
- снижение уровня памяти;
- частичная потеря звуковосприятия;
- апатия к происходящему вокруг, потеря активности;
- отсутствие сексуального влечения;
- трансформация поведенческих реакций;
- иногда возникает беспричинная эйфория или паническая атака, бред или галлюцинации;
- частичная потеря координации;
- забывание слов, невозможность выполнить простейшие математические вычисления в уме.

При дальнейшем течении болезни происходит развитие последующей дегенеративной симптоматики:
- периодическая парализация мышечного аппарата;
- эпилептические припадки;
- сильная дрожь в конечностях;
- нервные подергивания отдельных мышечных групп, чаще всего носогубного треугольника и глазных век;
- прогрессирующее слабоумие, потеря мыслительных способностей;
- речевая дисфункция вплоть до полного распада осознанного звуковоспроизведения;
- у пациента пропадает чувствительность к внешнему воздействию.
На завершающей стадии недуга наблюдается:
- глубинная деменция;
- полная отрешенность, отсутствие контактности;
- потеря контроля функций выделения, больному требуется постоянный присмотр и уход;
- атрофия мышечного аппарата, дегенерация безусловных рефлексов (например, глотания);
- впадение в коматозное состояние с последующим летальным исходом.
Диагностические меры
Основываясь на прогрессирующей симптоматике, лечащий врач – невролог обращается к инструментальным методикам диагностирования: ЭЭГ, ПЭТ, магнитно-резонансное сканирование мозга головы, пункцию мозговых тканей. При затруднениях в диагностировании может быть использована проникающая биопсия. Это самый информативный способ диагностирования при данном недуге, так как у специалистов появляется возможность выявить повышенное количество патогенного приона в изъятых на анализ тканях.
Лечение аномального состояния
Восстановление пораженных волокон в современной медицине в настоящее время недоступно. Медики могут назначить только симптоматическое лечение, направленное на купирование психических проявлений, противоэпилептические препараты, медикаменты, назначаемые при болезни Паркинсона. Лечение известными противовирусными веществами не достигает какого-либо эффекта, также как и предварительная вакцинация пациентов и домашнего скота. Некоторый эффект выявлен при применении Брефелдина А, который купирует разрастание агрессивных белков в пораженных нейронах, но данный эффект является временным. Полностью избавиться от аномалии еще не удалось, но подходы к лечению находятся в стадии активной разработки.
Прогноз и профилактика
Так как медицинскими способами невозможно купировать проявления недуга, а трансформация приона происходит с высокой скоростью, единственным финалом для заболевших пациентов, к сожалению, является летальный исход. Средняя продолжительность жизни после проявления первых признаков составляет 8-12 месяцев в 90% случаев. Лишь небольшая часть больных продолжают жить в течение 24-26 месяцев.
Профилактические меры заключаются в тщательной обработке медицинских инструментов перед оперативным вмешательством, анализе тканей, предназначенных для целей трансплантологии и отказе от мясной продукции от крупного рогатого скота. Самые частые случаи заболеваемости описанной патологией фиксируются в таких странах, как Великобритания, Чили, Словакия и Израиль.
Что такое болезнь Крейтцфельдта-Якоба?
Болезнь Крейтцфельдта-Якоба (болезнь Кройцфельдта-Якоба, БКЯ) — это редкое неврологическое заболевание (заболевание нервной системы), которое вызывает повреждение головного мозга.
Он относится к группе заболеваний, называемых трансмиссивными губчатыми энцефалопатиями, или прионными заболеваниями, которые поражают людей и животных.
БКЯ смертельна, а лечения никакого нет, если только симптоматическое. Возникает болезнь из-за неправильного белка, называемого прионом, загрязняющего нервную систему.

Прионы — это инфекционные частицы, состоящие из ненормально сложенного белка. В некоторых отношениях прион похож на вирус, так как он может размножаться и вызывать заболевание. Но, в отличие от вируса, он сделан полностью из белка и не имеет генетического материала.
Это делает прионы намного жестче, чем вирусы или бактерии. Они могут выдерживать экстремальные температуры и радиацию, и устойчивы к расщеплению ферментами, обычно контролирующими уровень белка в организме. Антибиотики и противовирусные препараты на них не влияют.
Прионы убивают клетки головного мозга и создают в мозге отверстия, придавая губчатый вид.
Типы болезни Крейтцфельдта-Якоба
Существует 4 различных типа болезни Крейтцфельдта-Якоба, каждый из которых имеет свою причину:
- Спорадическая (sCJD) является наиболее распространенной формой, на которую приходится 85% случаев болезни. Причина неизвестна, но обычно она затрагивает людей старше 40 лет.
- Новый вариант (nvCJD) был впервые идентифицирован в 1996 году. nvCJD вызывается употреблением мяса крупного рогатого скота, инфицированного коровьим бешенством. nvCJD в основном поражает людей в возрасте от 20 лет.
- Ятрогенная форма (1CJD) — инфекция передается от человека с заболеванием посредством медицинского или хирургического лечения. В наши дни его случаи крайне редки.
- Наследственная форма (fCJD) — это редкая форма прионной болезни, вызванная наследованием дефектного гена, продуцирующим прионы.
Симптомы и стадии
Симптомы и течение болезни Крейтцфельдта-Якоба различаются в зависимости от типа. Тем не менее, все четыре типа болезни имеют некоторые схожие признаки, описанные ниже.
Болезнь Крейтцфельдта-Якоба обычно начинается с эмоциональных или поведенческих проблем, таких как депрессия, беспокойство или возбуждение.
Бред (сильные убеждения в вещах, явно не соответствующих действительности) и галлюцинации (видение или слышание вещей, которых нет) типичны для нового варианта болезни (nvCJD).
У пациентов также развиваются неврологические проблемы (влияющие на нервную систему), такие как боль или онемение в частях тела.
В течение нескольких недель человек быстро ухудшается, становится запутанным и испытывает потерю памяти (симптомы, типичные для слабоумия).
Наряду с растерянностью и потерей памяти, человек может потерять координацию и равновесие, и начать слепнуть.
Месяцы спустя больные не могут ходить, разговаривать и заботиться о себе. Они не знают своего окружения и у них развиваются резкие движения мышцами.
Три четверти людей со спорадической (классической) формой болезнью Крейтцфельдта-Якоба умирают в течение шести месяцев после постановки диагноза, часто от инфекции пневмонии. Другие умирают в течение нескольких недель.
Причины болезни Крейтцфельдта-Якоба
Болезнь вызывается инфекционным белком в мозге, называемым прионом.
Белки — это молекулы, состоящие из аминокислот, помогающие клеткам нашего организма функционировать.
Белки начинаются как цепочка аминокислот, которые затем складываются в трехмерную форму. Такое “складывание белка” (фолдинг белка) позволяет им выполнять полезные функции в наших клетках.
Прионные белки (которые не являются теми же самыми инфекционными прионами, которые вызывают БЯК, т.е. нормальные прионные белки) — это тип белков, встречающихся в мозгу и некоторых других тканях нервной системы. Точная роль прионных белков в мозгу неизвестна, но считается, что они могут иметь какое-то отношение к долгосрочной памяти.
Иногда ошибки происходят во время фолдинга белка, в результате прионный белок не может быть использован организмом. Эти неправильно уложенные прионные белки обычно перерабатываются организмом, но иногда они могут накапливаться, что может вызвать проблемы, такие как болезнь Альцгеймера.
Прионы — это неправильно свернутые прионные белки, которые проникают в клетки мозга и также приводят к неправильному складыванию нормальных белков. Это приводит клетки мозга к смерти, высвобождая больше прионов, заражая другие клетки головного мозга.
В конце концов, кластеры клеток головного мозга погибают и заменяются отложениями прионов, называемыми бляшками. Эти бляшки производят небольшие отверстия в мозге, придавая ему губчатый вид. Повреждение головного мозга вызывает умственные и физические нарушения и возможную смерть, связанную с болезнью.
Прионы могут жить в нервной ткани (в головном или спинном мозге), в течение очень долгого времени, даже после смерти человека или животного.
Спорадическая БЯК (sCJD) является наиболее распространенным типом болезни, хотя все еще очень редко. Неизвестно, что вызывает sCJD, но она может быть связано с тем, что нормальный белок самопроизвольно превращается в прион или нормальный ген самопроизвольно превращается в дефектный ген, продуцирующий прионы. sCJD обычно поражает людей старше 40 лет.

Существуют явные доказательства того, что новый вариант БЯК (vCJD) вызван тем же штаммом прионов, что и губчатая энцефалопатия крупного рогатого скота (также известная как коровье бешенство).
Исследование правительства Великобритании в 2000 году пришло к выводу, что прион распространялся через крупный рогатый скот, которого кормили мясокостной смесью, содержащей следы зараженного мозга или спинного мозга. Затем прион оказался в переработанных мясных продуктах, таких как бифбургеры, и вошел в пищевую цепь человека.
Ятрогенная форма болезни является результатом передачи инфекции от человека с БЯК посредством медицинского или хирургического лечения.
Большинство ятрогенных случаев болезни Крейтцфельдта-Якоба (1CJD) произошло при использовании гормона роста человека, который использовался для лечения детей с ограниченным ростом. Между 1958 и 1985 годами тысячи детей лечились гормоном, который в то время был извлечен из гипофиза (железа, находящегося у основания черепа) человеческих трупов. У крошечного меньшинства этих детей развился БЯК, поскольку полученные ими гормоны были взяты из желез, инфицированных людей.
Несколько других случаев 1CJD произошли, когда люди получили пересадку зараженной ткани или вступили в контакт с хирургическими инструментами, которые были заражены болезнью Крейтцфельдта-Якоба. Это произошло из-за того, что прионы жестче, чем вирусы или бактерии, поэтому нормальный процесс стерилизации хирургических инструментов не работал.
Эта очень редкая форма болезни, вызвана она наследственной мутацией (дефектом) гена, который производит нормальные белки. Похоже, что измененный ген продуцирует прионы, вызывающие БЯК.
У каждого есть две копии своих генов, но мутированный ген является доминирующим. Это означает, что человеку нужно унаследовать только один мутированный ген для развития болезни.
Теоретически, болезнь может передаваться от больного человека другим, но только через инъекцию или потреблении мозговой ткани.
Считается, что болезнь Крейтцфельдта-Якоба не передается при обычном повседневном контакте с больными или воздушно-капельным путем, кровяным контактом или половым контактом.
Диагностика
Диагноз обычно основывается на истории болезни, симптомах и серии анализов (см. ниже). Невролог (врач, специализирующийся на состояниях нервной системы) проведет тесты, чтобы исключить другие состояния с похожими симптомами, такие как болезнь Альцгеймера, болезнь Паркинсона или опухоль головного мозга.
Единственный способ подтвердить диагноз болезни Крейтцфельдта-Якоба — это непосредственно исследовать ткань головного мозга с помощью биопсии головного мозга (см. ниже) или, если человек умер, после вскрытия.
Клинический невролог исключит другие заболевания с похожими симптомами и проверит наличие некоторых общих признаков болезни, выполнив любой из следующих тестов:
- Магнитно-резонансная томография (МРТ) головного мозга. При ней используются сильные магнитные поля и радиоволны для получения детального изображения мозга, и могут обнаруживаться отклонения, характерные для БЯК.
- Электроэнцефалограмма (ЭЭГ). Исследование регистрирует активность мозга и может обнаруживать аномальные электрические паттерны, наблюдаемые при спорадической форме БЯК.
- Люмбальная пункция. В нижнюю часть позвоночника вводится игла для отбора пробы спинномозговой жидкости (которая окружает головной и спинной мозг) для исследования в лаборатории. Если в жидкости обнаруживается белок, называемый 14-3-3, это указывает на то, что возможно человек болен болезнью Крейтцфельдта-Якоба (этот белок обнаруживается почти во всех случаях классической формы заболевания и в 50% случаев нового варианта болезни).
- Биопсия миндалин. Небольшой кусочек ткани берется из миндалин и проверяется на наличие аномальных прионов, обнаруживаемых при новом варианте БЯК (их нет в других типах).
- Генетический тест. Это простой анализ крови, показывающая есть ли мутация (ошибка) в гене, который производит нормальный белок. Положительный результат может указывать на наследственную форму заболевания.
Во время биопсии головного мозга хирург просверливает крошечное отверстие в черепе и удаляет маленький кусочек мозговой ткани с помощью очень тонкой иглы. Это делается под общим наркозом (человек спит).
Поскольку биопсия головного мозга несет в себе риск вызвать повреждение головного мозга или судороги (эпилепсию), она проводится только в нескольких случаях, когда существует опасение, что у пациента нет болезни Крейтцфельдта-Якоба, но есть какое-то другое излечимое состояние.
Лечение
Не существует доказанной терапии или лечения какой-либо формы болезни Крейтцфельдта-Якоба.
Множество потенциальных терапевтических вмешательств при болезни остаются в настоящее время на уровне дискуссии.
Лечение включает попытки сохранить человеку максимально комфортно короткую жизнь и уменьшить симптомы с помощью лекарств. Например:
- Психологические симптомы, такие как беспокойство и депрессия, можно лечить с помощью седативных средств и антидепрессантов.
- Другие лекарства, такие как клоназепам и вальпроат натрия, могут быть использованы для лечения мышц и тремора.
- Обезболивающие препараты на основе опиатов могут обеспечить эффективное обезболивание.
Когда кому-то ставят диагноз БЯК, его направляют в соответствующий центр для диагностики и лечения. Врач и медсестра из этих служб будут назначены пациенту для связи с местными службами, такими как врач общей практики, социальный работник, физиотерапевт и специалист по трудотерапии.
Доступны команды специалистов для диагностики и оказания клинической и эмоциональной поддержки пациентам и их семьям, а также работа вместе с местной командой по уходу. В местную команду по уходу могут входить врачи и медсестры, специалисты по гигиене труда, диетологи, консультанты по вопросам недержания и социальные работники.
По мере прогрессирования заболевания людям с болезнью Крейтцфельдта-Якоба потребуется значительный уход и практическая поддержка.
Помимо помощи в кормлении, мытье и передвижении, некоторые люди могут нуждаться в помощи при мочеиспускании. Часто требуется использование катетера (трубки, которая вставляется в мочевой пузырь и используется для слива мочи).
У многих людей возникают проблемы с глотанием, поэтому им, возможно, придется питаться и пить через трубку для кормления.
Может быть возможно лечить людей с БЯК и в домашних условиях, но это будет зависеть от прогрессирования и тяжести заболевания.
Давление при заботе о ком-то с БЯК может быть трудным. Вместо того, чтобы справляться с проблемой в домашних условиях, многие лица, осуществляющие уход, предпочитают пользоваться услугами больниц.
Прогноз
Не существует лекарство от БКЯ. Болезнь быстро ухудшает состояние человека до такой степени, что он больше не может заботиться о себе и не может двигаться или говорить.
Большинство людей со спорадической формой БКЯ умирают в течение 6 месяцев после постановки диагноза, часто от пневмонии. Люди с новым вариантом болезни живут в среднем чуть больше года.
Профилактика
Поскольку связь между коровьем бешенством и болезнью Крейтцфельдта-Якоба была подтверждена, предупредить развитие заболевания поможет качественная термическая обработка мяса животных, а также повышение иммунной системы организма.
Читайте также:
