
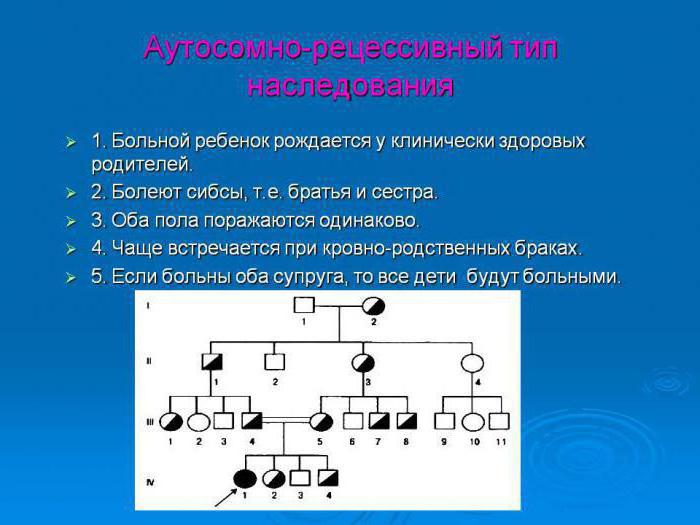
Аутосомно-доминантный тип наследования онихомикоза
Чаще всего патологии передает тип наследования аутосомно-доминантный. Это моногенное наследование одного из признаков. Помимо этого, болезни могут передаваться детям аутосомно-рецессивным и аутосомно-доминантным типом наследования, а также по митохондриальному признаку.

Типы наследования
Моногенное наследование гена может быть рецессивным и доминантным, митохондриальным, аутосомным или сцепленным с половыми хромосомами. При скрещивании может получиться потомство с самыми разными типами признаков:
- аутосомно-рецессивными;
- аутосомно-доминантными;
- митохондриальными;
- Х-доминантное сцепление;
- Х-рецессивное сцепление;
- У-сцепление.
Разные типы наследования признаков - аутосомно-доминантный, аутосомно-рецессивный и другие, способны передавать разным поколениям мутантные гены.
Особенности наследования аутосомно-доминантного типа
Аутосомно-доминантный тип наследования заболевания характеризуется передачей мутантного гена в гетерозиготном состоянии. У потомства, получившего мутантный аллель, может проявиться генное заболевание. При этом вероятность проявления измененного гена у мужчин и женщин одинаковая.

При проявлении у гетерозигот признак наследования не оказывает серьезного влияния на здоровье и функцию к воспроизводству. Гомозиготы с мутантным генном, который передал тип наследования аутосомно-доминантный, как правило, нежизнеспособны.
У родителей мутантный ген располагается в половой гамете совместно со здоровыми клетками, и вероятность его получения у детей будет равняться 50 %. Если доминантный аллель будет изменен не полностью, то дети таких родителей будут полностью здоровы на генном уровне. При низком уровне пенентрантности мутантный ген может проявляться не в каждом поколении.
Чаще всего тип наследования аутосомно-доминантный передает заболевания из поколения в поколение. При этом виде наследования у больного ребенка один из родителей болеет тем же заболеванием. Однако, если в семье болеет только один родитель, а второй имеет здоровые гены, то дети могут и не получить по наследству мутантный ген.
Пример наследования по аутосомно-доминантному типу
Тип наследования аутосомно-доминантный может передавать более 500 разных патологий, среди них: синдром Марфана, Элерса-Данло, дистрофия, болезнь Реклингхайзена, Гентингтона.
При изучении родословной можно проследить аутосомно-доминантный тип наследования. Примеры этому могут быть разные, но самый яркий – это болезнь Гентингтона. Она характеризуется патологическими изменениями нервных клеток в структурах переднего мозга. Проявляется недуг забывчивостью, слабоумием, проявлением непроизвольных телодвижений. Чаще всего это заболевание проявляется после 50 лет.

При прослеживании родословной можно выяснить, что хотя бы один из родителей страдал такой же патологией и передал ее по аутосомно-доминантному типу. Если же у больного есть брат или сестра единокровные, но у них нет проявления болезни, значит, родители передали патологию по гетерозиготному признаку Аа, при котором генные нарушения встречаются у 50 % детей. Следовательно, у потомства больного также могут родиться 50 % детей с видоизмененным геном Аа.
Аутосомно-рецессивный тип
При аутосомно-рецессивном наследовании отец и мать являются носителями патогена. У таких родителей 50 % детей рождаются носителями, 25 % - здоровыми и столько же - больными. Вероятность передачи патологического признака девочкам и мальчикам одинакова. Однако заболевания аутосомно-рецессивного характера могут передаться не каждому поколению, а проявляться через одно-два поколения потомства.
Примером заболеваний, передающихся по аутосомно-рецессивному типу, могут быть:
- болезнь Тоя-Сакса;
- нарушения обмена веществ;
- муковисцидоз и пр.
При обнаружении детей с аутосомно-рецессивным типом генных патологий выясняется, что родители находятся в родственной связи. Такое часто наблюдается в закрытых поселениях, а также в местах, где разрешаются кровные браки.
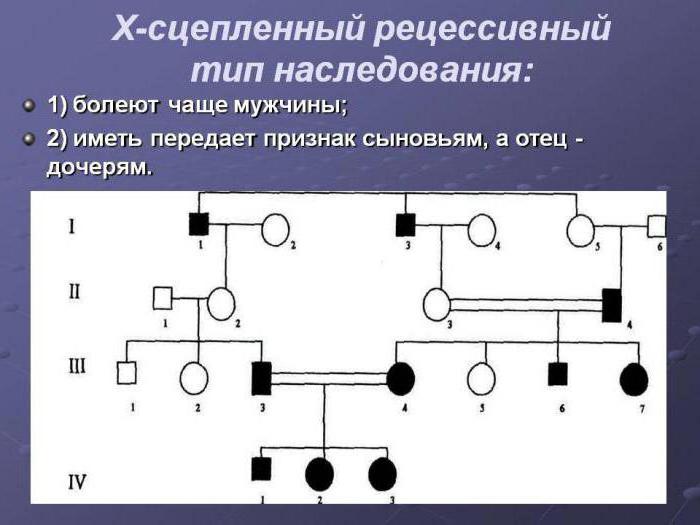
Х-хромосомное наследование
Х-ромосомный тип наследования у девочек и мальчиков проявляется по-разному. Это обусловлено наличием сразу двух Х-хромосом у женщины и одной у мужчины. Женский пол получает свои хромосомы по одной от каждого родителя, а мальчики – только от матери.
По этому типу наследования чаще всего патогенный материал передается женщинам, так как у них больше вероятности получить патогены от отца или матери. Если же носителем доминантного гена в семье является отец, то все мальчики будут здоровы, а у девочек будет проявляться патология.
При рецессивном типе Х-сцепления хромосом болезни проявляются у мальчиков с гемизиготным типом. Женщины всегда будут носителями больного гена, так как они гетерозиготные (в большинстве случаев), но если у женского пола будет гомозиготный признак, то она может заболеть.
Примером патологий с рецессивной Х-хромосомой могут быть: дальтонизм, дистрофия, болезнь Хантера, гемофилия.
Митохондриальный тип
Данный тип наследования является относительно новым. Митохондрии передаются с цитоплазмой яйцеклетки, в которых более 20 000 митохондрий. Каждая из них содержит хромосому. При данном типе наследования патологии передаются только по материнской линии. От таких матерей все дети рождаются больными.
При проявлении митохондриального признака наследственности у мужчин рождаются здоровые дети, так как этот ген не может передаваться от отца к ребенку, поскольку в спермии нет митохондрий.
Наша команда профессионалов ответит на ваши вопросы
Данная брошюра содержит информацию о том, что такое доминантный тип наследования и каким образом наследуются доминантные заболевания. Для того, чтобы лучше понять особенности доминантного наследования, вначале будет полезно узнать, что такое гены и хромосомы.
Наше тело состоит из миллионов клеток. Большинство клеток содержат полный набор генов. У человека тысячи генов. Гены можно сравнить с инструкциями, которые используются для контроля роста и согласованной работы всего организма. Гены отвечают за множество признаков нашего организма, например, за цвет глаз, группу крови или рост.
Гены расположены на нитевидных структурах, называемых хромосомами. В норме, в большинстве клеток организма содержится по 46 хромосом. Хромосомы передаются нам от родителей – 23 от мамы, и 23 от папы, поэтому мы похожи на своих родителей. Таким образом, у нас два набора по 23 хромосомы, или 23 пары хромосом. Так как на хромосомах расположены гены, мы наследуем по две копии каждого гена, по одной копии от каждого из родителей. Хромосомы (следовательно, и гены) состоят из химического соединения, называемого ДНК.
Иногда в одной копии гена возникает изменение (мутация), которое нарушает нормальную работу гена. Такая мутация может привести к развитию генетического (наследственного) заболевания, так как измененный ген не выполняет нужную для организма функцию.
Рисунок 1: Гены, хромосомы и ДНК

Рисунок 2: Как доминантные заболевания передаются от родителя к ребенку

Если у одного из родителей присутствует измененная копия гена, то он может передать ребенку либо нормальную копию, либо измененную. Таким образом, каждый из детей такого родителя будет иметь вероятность 50% наследования измененной копии и, следовательно, иметь генетическое заболевание.
В то же время, каждый из детей имеет такой же шанс – 50% - получить от родителя нормальную копию гена. В этом случае ребенок не будет болен этим наследственным заболеванием и не сможет передать измененные копии никому из своих будущих детей.
Оба возможных варианта (исхода) происходят случайным образом. Процент риска остается одним и тем же при каждой беременности и одинаков как для мальчиков, так и для девочек.
При заболеваниях с поздним началом (проявляющимся уже во взрослом возрасте, например, наследственная форма рака груди или хорея Гентингтона) люди могут умереть раньше начала манифестации наследственного заболевания от совсем других причин, и наследственное заболевание не успевает проявить себя. Однако родители могли передать заболевание своим детям.
Иногда пациент с доминантным заболеванием может оказаться первым больным в семье. Это может объясняться тем, что в сперматозоиде или яйцеклетке, из которых развился данный ребенок, произошла новая мутация (изменение) в гене, впервые в поколениях семьи. Если такое происходит, то родители этого пациента здоровы. В этом случае вероятность рождения у этих родителей другого ребенка с таким же заболеванием очень мала, однако этот вопрос обязательно следует обсудить с врачом. Однако, больной ребенок (как сын, так и дочь), у которого появился измененный ген, в будущем может передать его своим детям.
Если у кого-либо в семье есть доминантное заболевание, возможно, Вы будете обсуждать это с другими членами семьи. Эта информация может помочь другим членам семьи для решения вопроса об обследовании и диагностике заболевания. Это может быть особенно важно для тех членов семьи, у которых уже есть или будут дети.
Некоторым людям может оказаться сложно обсуждать свое генетическое заболевание с другими членам семьи. Они могут бояться причинить беспокойство членам семьи. В некоторых семьях люди из-за этого испытывают сложности в общении и теряют взаимопонимание с родственниками. Врачи-генетики, как правило, имеют большой опыт в решении подобных семейных ситуаций и могут помочь Вам в обсуждении проблемы с другими членами семьи.
Все характерные признаки нашего организма проявляются под действием генов. Иногда за это отвечает только один ген, но чаще всего бывает, что сразу несколько единиц наследственности несут ответственность за проявление того или иного признака.
Уже научно доказано, что для человека проявление таких признаков, как цвет кожи, волос, глаз, степень умственного развития, зависит от деятельности сразу множества генов. Это наследование совсем не в точности подчиняется законам Менделя, а выходит далеко за его рамки.
Изучение генетики человека не только интересно, но и важно с точки зрения понимания наследования различных наследственных заболеваний. Сейчас уже достаточно актуальным становится обращение молодых пар в генетические консультации, чтобы, проанализировав родословную каждого супруга, можно было бы с уверенностью утверждать, что ребенок родится здоровым.
Типы наследования признаков у человека
Если знать, как наследуется тот или иной признак, то можно предсказать вероятность его проявления у потомства. Все признаки в организме можно разделить на доминантные и рецессивные. Взаимодействие между ними не такое уж и простое, и порой недостаточно знать, какой из них относится к какой категории.
Сейчас в научном мире существуют следующие типы наследования у человека:
- Моногенное наследование.
- Полигенное.
- Нетрадиционное.
Эти типы наследования в свою очередь также подразделяются на некоторые разновидности.
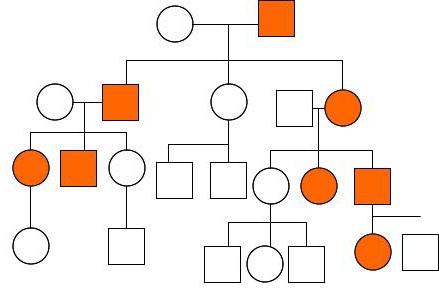
В основе моногенного наследования лежат первый и второй законы Менделя. Полигенное основано на третьем законе. При этом подразумевается наследование нескольких генов, чаще всего неаллельных.
Нетрадиционное наследование не подчиняется законам наследственности и осуществляется по своим, никому не известным, правилам.
Моногенное наследование
Эта разновидность наследования признаков у человека подчиняется менделеевским законам. Учитывая тот факт, что в генотипе присутствует по две аллели каждого гена, взаимодействие между женским и мужским геномом рассматривается отдельно для каждой пары.
Исходя из этого, выделяют следующие типы наследования:
- Аутосомно-доминантный.
- Аутосомно-рецессивный.
- Сцепленное с Х-хромосомой доминантное наследование.
- Х-сцепленное рецессивное.
- Голандрическое наследование.
Каждый тип наследования имеет свои особенности и признаки.
Признаки аутосомно-доминантного наследования
Тип наследования аутосомно-доминантный - это наследование преобладающих признаков, которые располагаются в аутосомах. Фенотипические проявления их могут сильно отличаться. У некоторых признак может быть едва заметным, а бывает и слишком интенсивное его проявление.
Тип наследования аутосомно-доминантный имеет следующие признаки:
- Больной признак проявляется в каждом поколении.
- Количество больных и здоровых примерно одинаковое, их соотношение 1:1.
- Если дети у больных родителей рождаются здоровыми, то и их дети будут здоровы.
- Болезнь одинаково затрагивает как мальчиков, так и девочек.
- Заболевание одинаково передается от мужчин и женщин.
- Чем сильнее влияние на репродуктивные функции, тем больше вероятность появления различных мутаций.
- Если оба родителя больны, то ребенок, рождаясь гомозиготой по этому признаку, болеет более тяжело по сравнению с гетерозиготой.
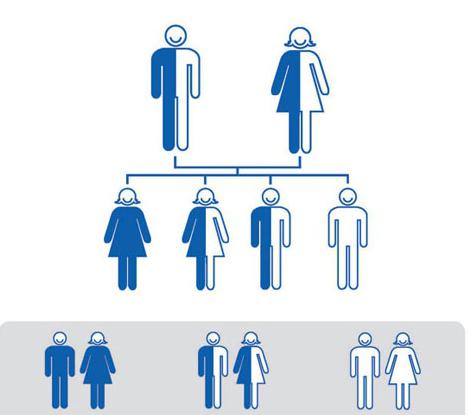
Все эти признаки реализуются только при условии полного доминирования. При этом только присутствия одного доминантного гена будет достаточно для проявления признака. Тип наследования аутосомно-доминантный можно наблюдать у человека при наследовании веснушек, курчавых волос, карих глаз и многих других.
Аутосомно-доминантные признаки
Большая часть лиц, которые являются носителями аутосомно-доминантного патологического признака, являются по нему гетерозиготами. Многочисленные исследования подтверждают, что гомозиготы по доминантной аномалии имеют более серьезные и тяжелые проявления по сравнению с гетерозиготами.
Этот тип наследования у человека характерен не только для патологических признаков, но и некоторые вполне нормальные так наследуются.
Среди нормальных признаков с таким типом наследования можно отметить:
- Вьющиеся волосы.
- Темные глаза.
- Прямой нос.
- Горбинка на переносице.
- Облысение в раннем возрасте у мужчин.
- Праворукость.
- Способность сворачивать язык трубочкой.
- Ямочка на подбородке.

Среди аномалий, которые имеют тип наследования аутосомно-доминантный, наиболее известны следующие:
- Многопалость, может быть как на руках, так и на ногах.
- Сращение тканей фаланг пальцев.
- Брахидактилия.
- Синдром Марфана.
- Близорукость.
Если доминирование неполное, то проявление признака можно наблюдать не в каждом поколении.
Аутосомно-рецессивный тип наследования
Проявиться признак при этом типе наследования может только в случае образования гомозиготы по этой патологии. Такие болезни протекают более тяжело, потому что обе аллели одного гена имеют дефект.
Вероятность проявления таких признаков повышается при близкородственных браках, поэтому во многих странах союз между родственниками заключать запрещено.
К основным критериям такого наследования можно отнести следующие:
- Если оба родителя здоровы, но являются носителями патологического гена, то ребенок будет болен.
- Пол будущего ребенка не играет при наследовании никакой роли.
- У одной семейной пары риск рождения второго ребенка с такой же патологией составляет 25%.
- Если посмотреть родословную, то прослеживается горизонтальное распределение больных.
- Если оба родителя больны, то все дети будут рождаться с такой же патологией.
- Если один родитель болен, а второй является носителем такого гена, то вероятность рождения больного ребенка составляет 50%
По такому типу наследуются очень многие заболевания, касающиеся обмена веществ.
Тип наследования, сцепленный с Х-хромосомой
Это наследование может быть как доминантным, так и рецессивным. К признакам доминантного наследования можно отнести следующие:
- Могут поражаться оба пола, но женщины в 2 раза чаще.
- Если болен отец, то он может передать больной ген только своим дочерям, потому что сыновья от него получают У-хромосому.
- Больная мать с одинаковой вероятностью награждает таким заболеванием детей обоего пола.
- Тяжелее протекает заболевание у мужчин, потому что у них отсутствует вторая Х-хромосома.

Если в Х-хромосоме находится рецессивный ген, то наследование имеет следующие признаки:
- Больной ребенок может родиться и у фенотипически здоровых родителей.
- Чаще всего болеют мужчины, а женщины являются носительницами больного гена.
- Если болен отец, то за здоровье сыновей можно не переживать, от него они не могут получить дефектный ген.
- Вероятность рождения больного ребенка у женщины-носительницы составляет 25%, если речь идет о мальчиках, то она повышается до 50%.
Так наследуются такие заболевания, как гемофилия, дальтонизм, мышечная дистрофия, синдром Калльмана и некоторые другие.
Аутосомно-доминантные болезни
Для проявления таких заболеваний достаточно наличия одного дефектного гена, если он является доминантным. Аутосомно-доминантные заболевания обладают некоторыми характеристиками:
- В настоящее время насчитывается около 4000 тысяч таких болезней.
- Поражаются одинаково особи обоих полов.
- Ярко проявляется фенотипический деморфизм.
- Если происходит мутация доминантного гена в гаметах, то она наверняка проявится в первом поколении. Уже доказано, что у мужчин с возрастом повышается риск получения таких мутаций, а, значит, своих детей они могут наградить такими заболеваниями.
- Болезнь зачастую проявляется во всех поколениях.
Наследование дефектного гена аутосомно-доминантного заболевания никак не связано с полом ребенка и степенью развития этой болезни у родителя.
К аутосомно-доминантным заболеваниям относятся:
- Синдром Марфана.
- Болезнь Хантингтона.
- Нейрофиброматоз.
- Туберозный склероз.
- Поликистоз почек и многие другие.
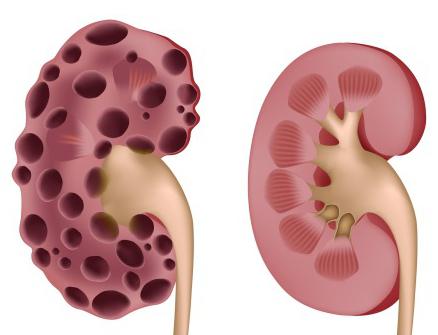
Все эти заболевания могут проявляться в разной степени у различных пациентов.
Синдром Марфана
Это заболевание характеризуется поражением соединительной ткани, а следовательно, и ее функционированием. Непропорционально длинные конечности с худыми пальцами дают основание предполагать синдром Марфана. Тип наследования этого заболевания - аутосомно-доминантный.
Можно перечислить следующие признаки этого синдрома:

Можно еще долго называть симптомы этого заболевания, но большая часть из них связана с костной системой. Окончательный диагноз будет поставлен после того, как пройдены все обследования, и характерные признаки обнаружены не менее чем в трех системах органов.
Можно отметить, что у некоторых признаки заболевания не проявляются в детском возрасте, а становятся очевидны несколько позже.
Даже сейчас, когда уровень медицины достаточно высок, полностью вылечить синдром Марфана невозможно. Используя современные препараты и технологии лечения, есть возможность продлить жизнь пациентов с таким отклонением и улучшить ее качество.
Самым главным аспектом в лечении является профилактика развития аневризмы аорты. Обязательны регулярные консультации кардиолога. В экстренных случаях показаны операции по пересадке аорты.
Хорея Хантингтона
Это заболевание также имеет тип наследования аутосомно-доминантный. Начинает проявляться с возраста 35-50 лет. Связано это с прогрессирующей гибелью нейронов. Клинически можно выявить следующие признаки:
- Беспорядочные движения в сочетании с пониженным тонусом.
- Асоциальное поведение.
- Апатия и раздражительность.
- Проявление шизофренического типа.
- Перепады настроения.
Лечение направлено только на устранение или снижение симптомов. Используют транквилизаторы, нейролептики. Никакое лечение не может остановить развитие заболевания, поэтому примерно через 15-17 лет после появления первых симптомов наступает смерть.
Полигенное наследование
Многие признаки и заболевания имеют тип наследования аутосомно-доминантный. Что это такое уже понятно, но в большинстве случаев все не так просто. Очень часто происходит наследование одновременно не одного, а сразу нескольких генов. Они проявляются в специфических условиях среды.
Отличительной особенностью такого наследования является возможность усиливать индивидуальное действие каждого гена. К основным признакам такого наследования можно отнести следующие:
- Чем тяжелее протекает болезнь, тем больше риск развития этого заболевания у родственников.
- Многие мультифакториальные признаки поражают определенный пол.
- Чем большее количество родственников имеет такой признак, тем выше риск появления этого заболевания у будущих потомков.
Все рассмотренные типы наследования относятся к классическим вариантам, но, к сожалению, очень многие признаки и болезни не поддаются объяснению, потому что относятся к нетрадиционному наследованию.
Планируя рождение малыша, не пренебрегайте посещением генетической консультации. Грамотный специалист поможет вам разобраться в вашей родословной и оценит риск рождения ребенка с отклонениями.
ГЕННЫЕ БОЛЕЗНИ
Генные болезни – это разнообразная по клинической картине группе заболеваний, обусловленная мутациями единичных генов.
При генных болезнях наблюдаются два вида изменения белковых продуктов:
· болезни, связанные с качественными изменениями белковых молекул
· заболевания характеризующиеся количественными изменениями содержания нормального белка в клетке (повышенное, пониженное), что вызвано чаще всего мутациями функциональных генов.
Как и для любой группы заболеваний, классификация генных болезней условна и многокомпонентна. По меньшей мере, три разных принципа могут быть положены в основу классификации генных болезней: генетический, клинический, патогенетический. В соответствии с генетическим принципом классификации генные болезни можно подразделить на группы согласно типам наследования:
· сцепленные с половыми хромосомами
Отнесение болезни к той или иной группе помогает врачу сориентироваться относительно ситуации в семье и определить вид медико-генетической помощи.
Аутосомно-рецессивные заболевания.
Известно более 1600 таких болезней. Аутосомно-рецессивные заболевания проявляются только у гомозигот, которые получили по одному рецессивному гену от каждого из родителей. Аутосомно-рецессивные болезни чаще всего обусловлены дефектами ферментов, реже - дефектами структурных белков. Именно поэтому многие врожденные нарушения обмена веществ попадают в эту группу болезней.
Нарушения аминокислотного обмена.
Самая многочисленная группа наследственных болезней обмена веществ. Почти все они наследуются по аутосомно-рецессивному типу.
Фенилкетонурия

— аутосомно-рецессивная болезнь аминокислотного обмена.
Частота в европейских странах 1:10000 новорожденных.
Эта болезнь характеризуется дефицитом печеночного фермента фенилаланингидроксилазы. Локус фенилкетонурии (фенилаланингидроксилазы) расположен в длинном плече 12-й хромосомы.
Ранняя диагностика фенилкетонурии и профилактическое лечение (искусственная диета) предупреждают развитие клинической картины болезни.
Дети, родившиеся с таким диагнозом, еще некоторое время назад были обречены на тяжелую умственную отсталость, поскольку поступающий с пищей фенилаланин не подвергался необходимым превращениям. В результате страдали функции мозга. Сейчас можно избежать столь тяжелых последствий, если сразу же после рождения больного ребенка исключить из пищи продукты, содержащие фенилаланин. Такие диеты разработаны и применяются. Диагностика фенилкетонурии у новорожденных не представляет трудностей, поэтому при всеобщем скрининге новорожденных и применении диетотерапии частота умственной отсталости вследствие фенилкетонурии может быть снижена. Диагноз ставится на основании клинической картины и результатов биохимического исследования мочи (фенилпировиноградная кислота) или крови (фенилаланинемия).
Нарушение липидного обмена.
Наследственные болезни обмена липидов - обширная группа различных по генезу состояний, патогенетически связанных с нарушением жирового обмена, в том числе - плазматические липидозы, для которых характерно накопление липидных субстанций внутри клеток.нарушения обмена липопротеидов.
Болезнь Тея-Сакса- это аутосомно-рецессивное генетическое заболевание, которое вызывает прогрессирующее ухудшение умственных и физических способностей ребенка. В основе заболевания лежит нарушение обмена ганглиозидов, сопровождающееся повышением их уровня в сером веществе мозга в 100-300 раз. Ганглиозиды накапливаются также в печени, селезенке. При болезни Тея-Сакса определяется дефицит гексозамининидазы А - одной из форм лизосомального фермента. Локализация гена - на 15-й хромосоме. Частота заболевания 1 : 250 000, среди евреев-ашкенази - 1 : 4000.
При рождении и в первые 3-4 мес жизни дети не отличаются от здоровых сверстников. Заболевание развивается медленно, ребенок становится менее активным, теряет приобретенные навыки, утрачивает интерес к игрушкам. Психомоторные нарушения начинают развиваться у детей с 4-6 мес. Дети становятся апатичными, перестают интересоваться окружающим, у них наблюдается мышечная гипотония. К концу 1 -го года жизни развивается слепота, обусловленная атрофией зрительных нервов; интеллект снижается до уровня идиотии. Постепенно развивается полная обездвиженность, наблюдаются судороги, не поддающиеся терапии. Смерть обычно наступает в возрасте 2-4 года.
Болезнь Гоше

Болезнь Гоше (керазиновый ретикулоэндотелиоз, цереброзидоклеточный липидоз) — редкое семейное заболевание, обусловленное накоплением в ретикулогистиоцитарных клетках глюкоцереброзида. В большинстве случаев болезнь наследуется по аутосомно-рецессивному, некоторых случаях — по доминантному типу. В общей популяции данная патология встречается у из 60 000 человек. Основные нарушения имеются на первом этапе утилизации глюкоцереброзида и обусловлены дефицитом глюкоцереброзидазы. Активность фермента при хронической форме болезни Гоше снижена на 65 %, а при острой — практически отсутствует.
Наиболее типичными проявлениями болезни Гоше служат увеличение размеров селезенки и печени, развитие анемии, тромбоцитопении, хронические боли в костях или развитие внезапных приступов сильнейших болей в костях (костные кризы). Последние сопровождаюются лихорадкой и местными островоспалительными явлениями (отек, покраснение), напоминающими картину остеомиелита. Реже болезнь может впервые проявиться переломом кости вследствие незначительной травмы. Поражение костей зачастую представляет основную клиническую проблему и может привести к тяжелой инвалидизации (обездвиженность вследствие многочисленных патологических переломов, деформации костей и суставов, необходимость замены разрушенных тазобедренных или плечевых суставов).
Выделяются три типа болезни Гоше:
- тип I – характеризуется отсутствием признаков поражения нервной системы;
- тип I I (острый нейронопатический) – встречается у детей раннего возраста и отличается тяжелым поражением головного мозга, больные редко доживают до возраста 2 лет;
- тип I I I (хронический нейронопатический) – объединяет более разнородную группу больных, у которых признаки поражения нервной системы могут проявляться как в раннем, так и в подростковом возрасте.
Тяжесть клинических признаков при болезни Гоше находится в прямой зависимости от энзимного дефекта. Чем больше снижена активность фермента, тем в более раннем возрасте и в более тяжелой форме проявляется заболевание.
Сфингомиелиноз (болезнь Нимана-Пика)

- внутриклеточный липидоз, характеризующийся накоплением в клетках ретикулоэндотелия фосфолипида сфингомиелина из-за нарушения активности сфингомиелиназы. Тип наследования - аутосомно-рецессивный. Частота заболевания составляет 1:10 000. Болезнь генетически гетерогенна: выделяются 4 варианта - А, В, С, D. При вариантах А и В отмечается дефицит сфингомиелиназы, при вариантах С и D активность энзима нормальная.
Заболевание встречается только в раннем детском возрасте и характеризуется злокачественным течением. Начало заболевания сопровождается отказом от еды, срыгиванием и рвотой. Затем выявляются задержка психомоторного развития и прогрессирующая гепатоспленомегалия. В дальнейшем развиваются спастический тетрапарез, глухота и слепота. Наблюдается умеренное генерализованное увеличение лимфатических узлов. Кожа приобретает коричневый оттенок. На глазном дне обнаруживается вишнево-красное пятно. При варианте болезни А (классическая форма) имеются тяжелые поражения нервной системы, которые сочетаются с выраженной гепатоспленомегалией. Вариант В отличается более поздними сроками манифестации (2- 6 лет), отсутствием неврологической симптоматики и хроническим течением. Чрезвычайной вариабельностью начала характеризуется вариант болезни С, при котором у 40% больных отмечается пролонгированная желтуха, исчезающая к 2-4 мес жизни, типична гепатоспленомегалия. Вариант D по клинической картине сходен с вариантом С, у больных часто наблюдаются миоклонические судороги.
Нарушение обмена гормонов.

Адреногенитальный синдром относится к группе наследственных нарушений биосинтеза стероидных гормонов. Известно несколько разновидностей наследственных дефицитов ферментов, обеспечивающих синтез стероидов. Ген стероид-21-гидроксилазы (СУР21В) локализован в коротком плече 6-й хромосомы. Частота дефицита 21-гидроксилазы - 1:5000 новорожденных. Недостаточность 21-гидроксилазы определяет до 90% всех случаев врожденной гиперплазии надпочечников. Известны два классических варианта этой болезни - сольтеряющая и простая вирильная форма. Другие формы встречаются значительно реже.
Сольтеряющая форма характеризуется полным дефицитом фермента и проявляется нарушением солевого обмена (дефицит минералокортикоидов). В патологический процесс вовлекается ренин-ангиотензинная система. У новорожденного отмечаются срыгивание, рвота, симптомы недостаточности периферического кровообращения, сонливость, потеря массы тела. Обезвоживание вызывает повышенную жажду. При биохимических исследованиях выявляются гиперкапиемия, гипонатриемия, ацидоз.
Простая вирильная форма характеризуется прогрессирующей вирилизацией, ускоренным соматическим развитием, повышенной экскрецией гормонов коры надпочечников. У новорожденных девочек при кариотипе 46,XX отмечается различная степень маскулинизации (от умеренной гипертрофии клитора до полного срастания губно-мошоночных складок с формированием мошонки и пениса). У мальчиков заболевание проявляется на 5-7-м году жизни преждевременным половым развитием.
ГЕННЫЕ БОЛЕЗНИ
Генные болезни – это разнообразная по клинической картине группе заболеваний, обусловленная мутациями единичных генов.
При генных болезнях наблюдаются два вида изменения белковых продуктов:
· болезни, связанные с качественными изменениями белковых молекул
· заболевания характеризующиеся количественными изменениями содержания нормального белка в клетке (повышенное, пониженное), что вызвано чаще всего мутациями функциональных генов.
Как и для любой группы заболеваний, классификация генных болезней условна и многокомпонентна. По меньшей мере, три разных принципа могут быть положены в основу классификации генных болезней: генетический, клинический, патогенетический. В соответствии с генетическим принципом классификации генные болезни можно подразделить на группы согласно типам наследования:
· сцепленные с половыми хромосомами
Отнесение болезни к той или иной группе помогает врачу сориентироваться относительно ситуации в семье и определить вид медико-генетической помощи.
Аутосомно-доминантные заболевания
Известно более 3700 таких болезней. Как правило, они обусловлены дефектами структурных белков или нарушениями регуляции экспрессии (выраженности)генов.
Эти болезни поражают мужчин и женщин с одинаковой частотой. Исключение составляют аутосомные дефекты, наследование которых зависит от пола. Для аутосомно-доминантных болезней характерен фенотипический полиморфизм ( внутри одной семьи).
Синдром Марфана (СМ)

Синдром Марфана - наследственная доминантная болезнь соединительной ткани.
Причиной синдрома Марфана являются мутации в гене фибриллина (локализация в хромосоме 15q).
Наиболее специфичными для синдрома Марфана являются нарушения скелета, вывих хрусталика, сердечно-сосудистые изменения, эктазия твердой мозговой оболочки.
2. Глаза: вывих хрусталика, миопия, отслоение сетчатки, большая роговица, удлиненная ось глазного яблока, уплощение роговицы.
3. Сердечно-сосудистая система: аортальная регургитация, аневризма восходящей части аорты, расслоение аорты, митральная регургитация, застойные сердечные нарушения, пролапс митрального клапана, кальцификация митрального отверстия, аритмия.
4. Наружные покровы: паховые грыжи, атрофические стрии.
5. Легочная система: спонтанный пневмоторакс.
6. Нервная система: эктазия твердой мозговой оболочки, аномалии развития нервной системы.
При синдроме Марфана ростовые скачки и закрытие зон роста скелета наблюдаются на 2,4 года раньше у лиц мужского пола и на 2,2 года раньше у лиц женского пола. Рост взрослых мужчин равен в среднем 191 см, женщин - 175 см.
Частота синдрома Марфана в популяции равна 1:10 000- 1:15 000. Популяционных и этнических различий в частоте и клинической картине болезни не отмечено.
Нейрофиброматоз (болезнь Реклингхаузена).

Тяжелая многосистемная болезнь с аутосомно-доминантным типом наследования. Распространенность нейрофиброматоза I типа равна примерно 1:3500-5000 новорожденных, одинакова у обоих полов, у всех рас и этнических групп. Ген расположен на 17 хромосоме.
Симптоматика НФ разнообразна, в патологический процесс вовлекаются несколько систем. Диагноз НФ можно поставить при наличии у пациента не менее двух из перечисленных признаков, но при условии, что они не являются симптомами какой-нибудь другой болезни:
1) светло-коричневые пятна на коже; пигментные пятна появляются в детстве, и их число увеличивается с возрастом;
2) наличие двух и более нейрофибром любого типа или одной плексиформной нейрофибромы; их количество с возрастом увеличивается;
3) множественные, похожие на веснушки пигментные пятна в подмышечной ямке, паховой области, на других участках тела со складками;
4) костные изменения (дисплазия крыла крыловидной кости, врожденное искривление или утончение длинных трубчатых костей, ложный сустав);
5) глиома зрительного нерва; узелки Лиша (два и более) на радужной оболочке.
Лечение нейрофиброматоза I типа в основном хирургическое или симптоматическое. В настоящее время идет разработка патогенетической терапии (антифиброзные средства).
Серповидноклеточная анемия.

Характеризуется присутствием неправильных красных кровяных клеток (эритроцитов), устойчивой, серповидной формы. Ее наледование зависит от доминантного гена S, ответсвенного за синтез гемоглобина HbS. Симптомы:
-всегда наблюдается анемия, которая может привести к потере сознания, делает больного физически менее выносливым и может вызвать желтуху (связанную с чрезмерным распадом гемоглобина).
-первыми признаками серповидноклеточной анемии у младенца обычно являются опухание и болезненность кистей рук или стоп, слабость и искривление конечностей и иногда, несколько позднее, отказ от ходьбы.
-ребенок с серповидноклеточной анемией обычно выглядит бледным, возможно, слегка желтушным, но в остальных отношениях, как правило, здоров.
-у взрослых с серповидноклеточной анемией могут обнаруживаться симптомы хронической (постоянной или длительной) закупорки капилляров легких и почек, и может развиться хроническая легочная или почечная недостаточность. Эти два осложнения приводят к ранней смерти некоторых пациентов с серповидноклеточной анемией.
Патология зрения: катаракта и глаукома. Кактаракта – помутнение хрусталика. Глаукома – следствие повышенного внутриглазного давления. Оба заболевания приводят к слепоте, но в настоящее время излечиваются оперативным путем.
Патология скелета:брахидактилия - аномалия развития рук или ног, укорочение пальцев.
Полидактилиия - анатомическое отклонение, характеризующееся бо́льшим, чем в норме, количеством пальцев на руках или ногах у человека.
Синдактилия - проявляется в полном или неполном сращивании пальцев кисти или стопы в результате не наступившего их разъединения в процессе эмбрионального развития.
Ахондроплазия – общее заболевание костной системы: нарушение роста эпифизарных хрящей трубчатых костей, деформация основания черепа и носовых костей. Это приводит к развитию очень коротких конечностей при нормальном туловище и голове.
Последнее изменение этой страницы: 2017-01-26; Нарушение авторского права страницы
Читайте также:
