
Неонатальный гепатит с холестазом
Патофизиологические основы формирования
Мухина Ю.Г, Дегтярева А.В., Морозов И.А, Туманова Е.Л, Талалаев А.Г.
Кафедра детских болезней № 2 педиатрического факультета с курсом гастроэнтерологии и диетологии ФУВ, Кафедра патологической анатомии педиатрического факультета, РГМУ, ЦНИИГ.
Холестаз – нарушение образования и/или экскреции желчи по желчевыводящей системе, приводящее к внутриклеточному и/или внутрипротоковому накоплению компонентов желчи и проявляющееся желтухой, постоянной или периодической ахолией стула, темным цветом мочи, увеличением размеров печени, кожным зудом, повышением уровня прямой фракции билирубина, щелочной фосфатазы (ЩФ), гамма-глутаминтрансферазы (ГГТ), холестерина, бета-липопротеидов ( b -ЛПД) и желчных кислот (ЖК).
В норме секреция желчи зависит от активности ферментных и транспортных систем гепатоцитов и желчных протоков, а также от их структурного и функционального их взаимодействия. Образование первичных ЖК (холевой и хенодезоксихолевой) происходит исключительно в печени из холестерина. Образование вторичных ЖК – дезоксихолевой и литохолевой происходит вследствие 7 a -дегидроксилирования первичных под действием бактерий кишечника. Третичные ЖК и, в основном, урсодезоксихолевая синтезируются в печени путем изомеризации вторичных ЖК. Печеночно-кишечная циркуляция ЖК включает транспорт ЖК в крови, их захват гепатоцитом, транспорт внутри гепатоцита от синусоидального (базолатерального) полюса к канальцевому, конъюгацию деконъюгированных в кишечнике и ре-синтезированных ЖК, экскрецию их в кишечник и последующую реабсорбцию как конъюгированных, так и неконъюгированных ЖК.
Экскреторная функция является жизненно важной; нарушение которой приводит не только к патологическим изменениям гепатобилиарной системы, но и вовлекает в патологический процесс другие органы и системы. Синдром холестаза способствует реализации токсических эффектов различных компонентов желчи, из которых первостепенное значение имеют желчные кислоты, билирубин, холестерин, провоспалительные медиаторы и микроэлементы. Токсические эффекты компонентов желчи представлены в табл. 1.
Патологические эффекты компонентов желчи при синдроме холестаза.
Цитотоксичность, нарушение дыхательной цепи митохондрий, апоптоз, повреждение канальцев.
Нарушение структуры митохондрий
Повреждение клеточных мембран
Воспаление, гемодинамические эффекты
Перекисное окисление липидов
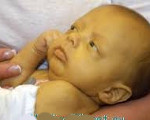
Дефицит желчи в кишечнике способствует дисрегуляции желудочно-кишечного тракта, нарушению процессов всасывания и, в первую очередь, жиров и жирорастворимых витаминов, нарушению функционального состояния поджелудочной железы. Отставание детей в физическом развитии является характерным признаком хронических холестатических заболеваний гепатобилиарной системы. Вторично формирующийся дисбаланс микроэлементов нарушает процессы оссификации костной системы с развитием остеопороза и метаболических нарушений сердечно-сосудистой системы. Например, при синдроме Алажиля длительно в течение нескольких лет существующий синдром холестаза с постоянно высоким уровнем холестерина и других липидов в крови способствует атеросклеротическим изменениям сосудов уже в детском возрасте. Высокий уровень сывороточных желчных кислот является ведущим фактором в развитии кожного зуда. Основные внепеченочные проявления длительно сохраняющегося синдрома холестаза представлены в таблице 2. Крайний вариант – на фотографии.
Внепеченочные проявления синдрома холестаза, фотография ребенка М.
Нарушение процессов всасывания

Отставание в физическом развитии
Признаки дефицита жирорастворимых витаминов:
Ксантомы, атеросклеротические изменения сосудов.
У новорожденных и детей первых месяцев жизни синдром холестаза является одним из ранних признаков широкого спектра заболеваний печени и желчевыводящих протоков. В генезе заболеваний лежит несостоятельность тех или иных структур гепатобилиарной системы, являющихся неотъемлемым звеном сложной системы образования и экскреции желчи. Эти дефекты могут быть генетически детерминированными или возникать под действием экзогенных факторов. В таблице 3 представлены заболевания гепатобилиарной системы в зависимости от уровня первичного поражения.
Первичное поражение клеток печени характерно для большинства гепатотропных инфекционных возбудителей, кроме того может наблюдаться при эндокринных и метаболических нарушениях, а также как проявление токсического действия лекарственных препаратов и осложнение длительного полного парентерального питания.
Гепатит, протекающий с синдромом холестаза, у новорожденных детей может быть вызван широким спектром инфекционных агентов: герпес-вирусом, цитомегаловирусом, энтеровирусами, вирусом краснухи, Эпштейна-Барра, гепатита В и С, возбудителем токсоплазмоза, листериоза, сифилиса и бактериальными агентами. В большинстве случаев неонатальный гепатит, вызванный вышеуказанными возбудителями, является одним из проявлений генерализованной инфекции. Выявление признаков инфекционного процесса и характерного для той или иной инфекции симптомокомплекса является необходимым условием для диагностики гепатита. Исключение составляют вирусные гепатиты В и С, при которых могут отмечаться изменения только печени. Однако у новорожденных детей гепатиты В и С встречаются редко.
Механизм патологического действия включает непосредственное повреждающее действие гепатотропных возбудителей с развитием специфического воспаления гепатобилиарной системы. При неонатальном гепатите синдром холестаза сочетается с биохимическим синдромом цитолиза и нарушением белок-синтетической функции печени. Характерно повышение неспецифических показателей воспаления в общем и биохимическом анализах крови. Наиболее объективным доказательством неонатального гепатита являются результаты морфологического исследования биоптата печени, при котором выявляются типичные для каждой инфекции признаки. Исследования, выявляющие возбудителя и/или антитела к нему подтверждают диагноз.

Рис. 1. Гистологическое исследование биоптата печени при ЦМВ-гепатите.
Среди метаболических заболеваний манифестирующих в течении первых месяцев жизни и проявляющихся синдромом холестаза следует отметить галактоземию, фруктоземию, тирозинемию, болезнь Нимана-Пика (тип С), дефицит- a -1-антитрипсина, неонатальный гемохроматоз и некоторые типы митохондриальной недостаточности с вовлечением ферментов цепи переноса электронов. В основе этих нарушений лежит генетически обусловленный дефект ферментной системы, приводящий к накоплению существующих в норме соединений, а также промежуточных продуктов их метаболизма, обладающих токсическим эффектом на клетки различных органов и являющихся ключевым патогенетическим фактором.
Наиболее часто встречается галактоземия, которая является наследственным заболеванием с аутосомно-рецессивным типом наследования, связанным с 9 хромосомой и включает патологические изменения со стороны печени, почек, ЦНС и глаз. Это заболевание встречается с частотой 1: 50.000 (1:18.000 -1:180.000) живорожденных новорожденных. Первые признаки заболевания появляются в период от нескольких часов до нескольких дней после начала энтерального питания молоком или смесью, содержащей галактозу. В основе этого заболевания лежит дефицит фермента галактоза-1-фосфатуридилтрансферазы, приводящий к накоплению галактозы и галактоза-1-фосфата. Галактоза превращается в галактитол, ответственный за развитие катаракты. Галактоза-1-фосфат значительно снижает синтез энергетических соединений (АТФ, ГТФ, ЦТВ), а также активность ферментов, принимающих участие в глюконеогенезе и синтезе глюкозы из гликогена, вызывая гипогликемию. Эти изменения вызывают тяжелые метаболические нарушения в клетках печени, головного мозга, почек и глаз, способствуют гемолизу эритроцитов. Галактоза-1-фосфат превращается в галактонат и галактонолактон. Эти метаболиты обладают непосредственным гепато-, нефро- и нейротоксическим действием. Описанные изменения лежат в основе характерного для галактоземии симптомокомплекса в виде нарушения общего состояния ребенка, проявляющегося срыгиваниями, рвотой, расстройством стула и др., признаками гемолитической анемии, патологическими изменениями со стороны печени в виде синдрома холестаза, синдрома цитолиза и нарушения синтетической функции. Первым клиническим признаком изменений со стороны ЦНС является внутричерепная гипертензия. Отмечаются также нарушения канальцевой функции почек и формирование катаракты. Подтверждением диагноза служит высокий уровень галактозы в крови, а также дефицит фермента галактоза-1-фосфатуридилтрансферазы.

Рис. 2. Гистологическое исследование биоптата печени при болезни Нимана-Пика, тип С.
Дефицит- a -1-антитрипсина отличается от описанных метаболических нарушений. Единственным его проявлением в неонатальном периоде является синдром холестаза; общее состояние ребенка, как правило, не страдает, патологические изменения в легких развиваются значительно позже, после 3-5 лет жизни. Заболевание имеет аутосомно-рецессивный тип наследования, связанный с нарушением одной аминокислотной цепочки гена, локализующегося в 14 хромосоме.
Конформационные изменения a 1-антитрипсина ( a 1-АТ) обуславливают его соединение с соответствующими ферментами. В связи с этим, те или иные структурные нарушения способствуют ослаблению или потере связывающей способности. Фенотипические варианты a 1-АТ отражают скорость его миграции в электрическом поле при pH между 4 и 5. Нормальный белок мигрирует в промежуточной, средней позиции и обозначается М ( medium ), белок, мигрирующий более медленно, обозначают Z или S ( slow ). В редких случаях отмечается нулевой фенотип - null , при котором полностью отсутствует белок. Наличие двух пар генов предполагает 6 основных вариантов фенотипа, отличающихся степенью выраженности дефицита: MM -100%, MS -75%, MZ -57%, SS -52%, SZ -37%, ZZ -16%. При Z -варианте отмечается замена глутамина на лизин в 342 положении, при S варианте глутамина на валин в 264 положении. Кроме того, при ZZ типе наряду с изменением аминокислотной последовательности имеет место нарушение эндоплазматического ретикулума гепатоцитов, что приводит к нарушению образования боковых углеводородных цепей. В связи с этим только 15% синтезированных полипептидов секретируется в плазму крови, тогда как 85% откладывается в эндоплазматическом ретикулуме, образуя глобулярные включения. Накопление белковых гранул при ZZ фенотипе является ведущим патогенетическим фактором патологических изменений печени. При S варианте отсутствуют патологические изменения гепатобилиарной системы, a -1-АТ не накапливается в клетках печени, однако он имеет нестабильную структуру и подвергается частичному внутриклеточному протеолизу, что обуславливает его сывороточный дефицит. Примерно в 21-50% случаев при неонатальном холестазе, обусловленном дефицитом a 1-АТ по ZZ фенотипу, отмечается развитие ювенильного цирроза печени. Наиболее типичным бронхолегочным проявлением заболевания, встречающимся при любом фенотипе у детей старше 3-5 летнего возраста, является эмфизема легких, однако дефицит a 1-АТ играет важную роль в патогенезе и других хронических заболеваний легких и, прежде всего бронхиальной астмы. Ориентировочным тестом, свидетельствующим о дефиците a -1-антитрипсина, является низкий сывороточный уровень a -1-глобулина. Подтвердить диагноз позволяет низкий уровень a -1-АТ наряду с выявлением характерных гистологических признаков в виде ШИК положительных включений, устойчивых к ферментативному перевариванию и располагающихся преимущественно в перипортальных зонах (рис. 3).

Рис. 3. Гистологическое исследование биоптата печени при дефиците a -1-АТ.
Канальцевая мембрана гепатоцита несет на своей поверхности транспортные системы, отвечающие за экскрецию различных компонентов желчи, органических и неорганических веществ, а также большинства ксенобиотиков. В генезе формирования синдрома холестаза играет роль генетически детерминированные дефекты 3-х транспортных систем канальцевой мембраны гепатоцита, лежащих в основе прогрессирующего семейного внутрипеченочного холестаза (ПСВХ) 3-х типов (табл. 4). Н аиболее часто встречается ПСВХ 1-го типа или болезнь Байлера, в основе которой лежит дефицит II -типа АТФ-азы, играющей ключевую роль в экскреции обоих первичных желчных кислот через канальцевую мембрану гепатоцита. При 11 типе ПСВХ отмечается нарушение экскреции преимущественно одной, хенодезоксихолевой кислоты через канальцевую мембрану гепатоцита, связанное с отсутствием на ее поверхности П-гликопротеина.
Типы и основные характеристики прогрессирующего семейного внутрипеченочного холестаза.
Молекулярные изменения канальцевой мембраны гепатоцита:
Холестаз новорожденного – это патологическое состояние, которое характеризуется нарушением секреции желчи, приводит к гипербилирубинемии и желтухе. Клинические проявления включают субиктеричность склер, пожелтение слизистых оболочек и кожи, гепатомегалию, задержку в физическом развитии, гиповитаминозы А, D, K, E. Диагностика холестаза новорожденного заключается в выявлении основного заболевания, лабораторной оценке работы печени, УЗИ, гепатобилиарном сканировании и биопсии печени. Лечение зависит от этиологического фактора и подразумевает рациональное питание, введение в рацион легкоусвояемых жиров, витаминотерапию или оперативное вмешательство.
МКБ-10
Общие сведения
Холестаз новорожденного – это гетерогенное состояние в педиатрии и неонатологии, которое проявляется нарушением выведения желчи с дальнейшим развитием гипербилирубинемии. Точную распространенность патологии установить тяжело, поскольку это состояние зачастую включают в структуру основного заболевания. Терапевтическая тактика и прогноз для жизни ребенка зависят от этиологического фактора холестаза новорожденного. Как правило, устранение ведущей патологии ведет к купированию холестатического синдрома. В тяжелых случаях может требоваться пересадка печени.
Причины
Все этиологические факторы холестаза новорожденного разделяют на внепеченочные и внутрипеченочные. Последняя группа включает в себя нарушения инфекционного, метаболического, генетического, аллоимунного или токсического происхождения.
- К инфекционным причинам относятся так называемые TORCH-инфекции: токсоплазмоз, краснуха, ЦМВ, герпес-вирус, сифилис, гепатит В и другие.
- Спровоцировать холестаз новорожденного могут такие нарушения метаболизма, как галактоземия, муковисцидоз, тирозинемия, недостаточность α1-антитрипсина.
- Генетическими факторами являются синдром Алажиля, кистозный фиброз, прогрессирующий семейный внутрипеченочный холестаз и др.
- Гестационное аллоиммунное поражение печени развивается при трансплацентарном прохождении материнских IgG в организм ребенка. Это вызывает активацию системы комплемента, которая способна повреждать гепатоциты.
- Длительное парентеральное вскармливание у тяжело недоношенных детей и наличие синдрома короткой кишки относятся к токсическим причинам холестаза новорожденного.
Перечень внепеченочных факторов развития холестаза новорожденного включает в себя все нарушения, которые ухудшают отток желчи из печени. Наиболее распространенная причина – атрезия желчевыводящих путей. Чаще всего наблюдается у недоношенных детей. Иногда, помимо внепеченочного билиарного тракта, поражаются и внутрипеченочные желчные протоки. Точная этиология данного состояния неизвестна. Предположительно, развитие билиарной атрезии вызывают инфекционные заболевания матери во время беременности.
Патогенез
Пусковым механизмом появления клинических симптомов при холестазе новорожденного является нарушение экскреции прямого билирубина, провоцирующее повышение его уровня в крови – гипербилирубинемию. Это приводит к дефициту желчных кислот в системном кровотоке. Так как они отвечают за всасывание и транспорт жиров и жирорастворимых витаминов (А, D, K, E) в организме ребенка возникает их дефицит. Результатом становится недостаточная прибавка массы тела, гиповитаминозы, задержка физического и психомоторного развития.
Симптомы холестаза новорожденного
Клиника холестаза новорожденного развивается на протяжении первых 10-14 дней жизни ребенка. Первичные проявления – беспокойство, снижение аппетита, недостаточная прибавка массы тела и роста. Почти сразу возникает субиктеричность склер и слизистых оболочек, при дальнейшем нарастании гипербилирубинемии отмечается желтушность кожных покровов.
На этом фоне моча приобретает коричневый оттенок, а кал становится бесцветным (ахолическим). Постепенно увеличивается печень, что проявляется незначительным увеличением живота. Также при холестазе новорожденного отмечается хронический зуд, клинические проявления гиповитаминозов А, D, K, E.
Осложнения
При дальнейшем прогрессировании основного заболевания, приводящего к фиброзу печени, развивается портальная гипертензия. Она, в свою очередь, становится причиной асцита и кровотечений из варикозно расширенных вен пищевода. Исход варьируется от длительного доброкачественного течения до быстрого развития печеночной недостаточности и цирроза с летальным исходом уже на первом году жизни.
Диагностика
Диагностика холестаза новорожденного включает в себя сбор анамнестических данных, физикальное обследование ребенка, лабораторные и инструментальные методы исследования. При опросе родителей педиатр или неонатолог обращают внимание на все возможные этиологические факторы: заболевания матери во время беременности, генетические патологии, врожденные аномалии развития ребенка. При физикальном осмотре определяется дефицит массы тела и роста, желтушность склер, слизистых оболочек и кожи, гепатомегалия. Подтверждающая диагностика:
- Лабораторные тесты. В биохимическом анализе крови выявляется гипербилирубинемия за счет прямой фракции, диспротеинемия. Возможно повышение трансаминаз – АлТ и АсТ. Характерные признаки холестаза новорожденного – ахолический, богатый на жиры кал и насыщенная уробилином моча. С целью исключения TORCH-инфекций проводится ИФА и бактериальный посев мочи. Также используются тесты на метаболические нарушения или генетические аномалии, которые могут вызывать развитие холестаза новорожденных – анализы на содержание NaCl в поте, галактозы в моче, уровень α1-антитрипсина в крови.
- Эхография. Первый этап – ультразвуковое исследование органов брюшной полости. Оно позволяет выявить гепатомегалию, аномалии строения желчного пузыря и желчевыводящих протоков. Далее выполняется УЗ-сканирование печени для оценки экскреции желчи на всех уровнях билиарного тракта – от внутрипеченочных протоков до двенадцатиперстной кишки.
- Биопсия печени. При невозможности подтвердить диагноз данными методами показана биопсия печени. При неонатальном холестазе гистологический анализ биоптата может выявить увеличенные портальные триады, пролиферацию и фиброзирование желчных протоков при атрезии желчевыводящий путей, гигантские полиядерные клетки при врожденном гепатите и др. При подозрении на аллоиммунное происхождение холестаза новорожденного определяется большое количество железа в гепатоцитах или тканях губ.
Лечение холестаза новорожденного
Терапия при холестазе новорожденного включает в себя несколько моментов: полноценное лечение основного заболевания, насыщение рациона ребенка витаминами А, D, K, E и среднецепочечными жирными кислотами, применение урсодезоксихолевой кислоты. Большое количество витаминов возмещает их недостаточное всасывание в кишечнике. Среднецепочечные жирные кислоты способны всасываться в кровоток даже в условиях дефицита желчных солей при холестазе новорожденного, тем самым компенсируя дефицит триглицеридов в организме. Урсодезоксихолевая кислота является гепатопротектором и желчегонным средством, стимулирующим работу печени и желчевыводящих путей и купирующим гипербилирубинемию.
При подозрении на аллоиммунное происхождение холестаза новорожденного (даже при неподтвержденном диагнозе) показано введение внутривенных иммуноглобулинов или обменное переливание крови.
При подтвержденной атрезии желчевыводящих путей уже на 1-2 месяце жизни проводится портоэнтеростомия по методу Касаи. Данная операция значительно улучшает показатель выживаемости, однако в будущем приводит к частым рецидивирующим холангитам, стойкому холестазу и задержке в психофизическом развитии. На фоне быстро развивающегося цирроза печени необходима трансплантация органа.
Прогноз и профилактика
Прогноз при холестазе новорожденного напрямую зависит от основного заболевания и эффективности терапевтических мероприятий. При атрезии желчевыводящих путей и отсутствии раннего хирургического вмешательства быстро развивается печеночная недостаточность. Далее это состояние трансформируется в цирроз с нарастающей гипертензией в системе воротной вены уже на первых месяцах жизни ребенка. Смерть наступает, как правило, в возрасте 10-12 месяцев. Инфекционные и метаболические причины холестаза новорожденного могут иметь как доброкачественный прогноз, так и быстро прогрессирующее течение. Аллоиммунное поражение печени на фоне отсутствия раннего лечения в большинстве случаев завершается летальным исходом.
Профилактика холестаза новорожденного включает в себя антенатальную охрану плода, медико-генетическое консультирование семейных пар и планирование беременности, регулярное посещение женской консультации, полноценное прохождение всех необходимых анализов и исследований. Данные меры позволяют оценить риск развития генетических заболеваний еще до начала беременности и выявить опасные заболевания уже на ранних сроках.

Определение
Гепатит - это воспалительное заболевание печени различной этиологии. Гепатит, симптомы которого развились у ребенка до 2-3-месячного возраста (чаще на первом месяце), считается неонатальным или перинатальным.
Причины
Гепатит - заболевание инфекционной природы. Причинами возникновения гепатита у новорожденных могут быть вирусы (вирус простого герпеса - ВПГ, цитомегаловирус - ЦМВ, Varicella-Zoster-вирус, другие герпес-вирусы, вирус гепатита В (ВГВ), вирус краснухи, ЕСНО- и Коксаки-вирусы и другие), Тreponema pallidum, бактерии (микобактерии туберкулеза, Listeria monocytogenes, кишечная палочка, стафилококк и другие бактерии-возбудители сепсиса), Toxoplasma gondii. Следует отметить, что передача вируса гепатита С от матери ребенку происходит достаточно редко, а гепатит при этом развивается не в первые месяцы жизни. Риск развития вирусного гепатита А у новорожденных также очень низкий. Вирус иммунодефицита человека (ВИЧ), который передается от матери ребенку, не вызывает гепатит. Однако у детей, рожденных ВИЧ-инфицированными матерями, значительно выше риск развития неонатального гепатита, чем в популяции, что объясняется иммунодефицитом у матери, наличием у нее оппортунистических и других инфекций, многие из которых также вызывают перинатальные инфекции (ВПГ, ЦМВ, ВГВ, сифилис, токсоплазмоз и другие), сопровождающиеся гепатитом.
Возбудитель попадает к плоду внутриутробно или во время родов от матери, у которой есть острый или хронический (персистирующий) инфекционный процесс, или латентное (бессимптомное) носительство микроорганизмов. Наиболее опасным для плода является первичное инфицирование матери во время беременности. Вторичная инфекция (реинфекция или реактивация хронической инфекции во время беременности) также может привести к инфицированию плода, но при этом риск перинатальной передачи возбудителя ниже, чем при первичном инфицировании. Медицинские манипуляции, ятрогенные вмешательства, патология беременности, нарушающие целостность плаценты, плодных оболочек, кожи плода также способствуют передаче возбудителя.
Симптомы
Клинические проявления гепатита выявляются чаще в первые недели жизни. Для гепатита характерны: гепатомегалия, желтуха, темная моча, обесцвеченный кал, явления кровоточивости, неспецифические симптомы инфекционного токсикоза. Желтуха, как правило, возникает после первых суток жизни. Цвет кожи варьирует от бледного с желтовато-зеленоватым оттенком до интенсивно желтого, чаще с зеленоватым оттенком. При некоторых инфекциях (например, ЦМВ-инфекции, ВПГ-инфекции, листериозе и других) на коже может выявляться различная сыпь - макулы, папулы, везикулы. Как результат гипопротеинемии, могут возникать отеки. К общим симптомам инфекционного токсикоза относят: снижение аппетита или отказ от еды, угнетение рефлексов периода новорожденности, вялость, метеоризм, рвота. Гепатомегалия (печень выступает из-под края реберной дуги более, чем на 3 сантиметра) может сопровождаться спленомегалией. Геморрагический синдром, полиорганная недостаточность, тяжелые метаболические нарушения являются непосредственными причинами смерти ребенка с неонатальным гепатитом.
Лабораторные признаки поражения печени:
- гипербилирубинемия за счет прямой и непрямой фракции - признак нарушения выделительной функции печени;
- повышение уровня трансаминаз (более специфична аланинаминотрансфераза, менее специфична аспартатаминотрансфераза) - признак цитолиза;
- повышение уровня щелочной фосфатазы и гамма-глутамил-транспептидазы - признак холестаза;
- гипокоагуляция и гипоальбуминемия - признак нарушения белково-синтетической функции печени.
Классификация
Неонатальные гепатиты делятся на три основных вида:
- Неонатальный гигантоклеточный гепатит врожденного характера с внутрипеченочным холестазом.
- Постнатальные гепатиты, которые обусловлены наличием внутриутробных инфекций, таких как листериоз, ЦВМ, токсоплазмоз и другие.
- Поражение печени токсико-септического характера при сепсисе и других инфекциях.
Диагностика
Дифференциальную диагностику неонатального гепатита проводят с наследственными нарушениями обмена веществ (галактоземия, тирозинемия), наследственными заболеваниями (дефицит α1-антитрипсина, муковисцидоз, неонатальный гемохроматоз, болезнь Байлера) и синдромами (Алажилля, Цельвегера), для которых также характерны гепатомегалия, гипербилирубинемия, повышение трансаминаз, нарушение функции печени. Дифференциальная диагностика неонатального гепатита инфекционной природы с наследственными заболеваниями очень важна, так как определяет правильную тактику ведения и прогноз.
Следует дифференцировать гепатит новорожденных и атрезию желчевыводящих путей (билиарную атрезию) - врожденную патологию, при которой желчевыводящие пути непроходимы или отсутствуют. При этом следует понимать, что билиарная атрезия может быть результатом гепатита плода, перенесенного во II триместре беременности. Реконструктивная хирургическая коррекция с созданием протоков (портоэнтеростомия, процедура Касаи) или пересадка печени являются способами лечения билиарной атрезии. Реконструктивную операцию желательно производить не позже, чем в двухмесячном возрасте, поскольку откладывание операции приведет к развитию цирроза, что резко снижает выживаемость детей. Даже при хирургическом вмешательстве вероятность смертельного исхода при билиарной атрезии - более 50%.
Профилактика
Не существует никакого специального лечения для неонатального гепатита. Многим детям назначают витаминные добавки, которые используются для контроля острых симптомов и стимуляции печени при выделении дополнительной желчи.
Гепатит новорожденных, вызванный вирусом гепатита А , обычно проходит сам в течение шести месяцев, но некоторые случаи являются результатом заражения вирусами гепатита В или С, что скорее всего, приводит к хроническому заболеванию печени. У детей, в конечном итоге, развивается цирроз и понадобится пересадка печени.
Дети с неонатальным гепатитом, вызванным цитомегаловирусом, краснухой или гепатитом A, B, C могут передавать инфекцию другим людям, которые вступают в тесный контакт с ребенком. Инфицированные дети, не должны вступать в контакт с беременными женщинами, потому что есть возможность того, что женщина может передавать вирус своему будущему ребенку.
Профилактику неонатальных гепатитов рекомендуется проводить путем вакцинации тех новорожденных детей, чьи матери имеют вирус гепатита С.
Холестаз - недостаточность секреции билирубина, приводящая к развитию конъюгированной гипербилирубинемии и желтухи.
Есть множество причин, выявляемых при лабораторных исследованиях, гепатобилиарном сканировании и иногда биопсии печени и хирургических вмешательствах. Лечение зависит от причины.
Причины неонатального холестаза у детей
Холестаз может возникнуть в результате вне-печеночных или внутрипеченочных нарушений, хотя некоторые состояния перекрываются. Наиболее распространенное расстройство - внепеченочная атрезия желчных путей. Существуют многочисленные внутрипеченочные нарушения, в общем называемые синдромом неонатально-го гепатита.
Заболевание редко возникает у недоношенных детей или у новорожденных при рождении.
Синдром неонатального гепатита (гигантоклеточный гепатит) представляет собой воспалительное заболевание печени новорожденных. Болезни обмена веществ включают недостаточность агантитрипсина, муковисцидоз, неонатальную болезнь накопления железа. Инфекционные причины - это врожденный сифилис, эхо-вирусы и некоторые герпесвирусы (ВПГ и цитомегаловирус); классические вирусы гепатита (А, В и С) являются менее общими причинами. Есть также ряд менее общих генетических дефектов, таких как синдром Алажилля.
При холестазе первичное нарушение затрагивает экскрецию билирубина, что приводит к избыточному содержанию билирубина в крови и снижению содержания солей желчных кислот в желудочно-кишечном тракте.
Симптомы и признаки неонатального холестаза у детей
Холестаз обычно выявляют в первые 2 нед жизни. Младенцы желтушные, часто имеют темный цвет мочи (содержит билирубин), ахолический стул и гепатомегалию. Если холестаз сохраняется, хронический зуд является обычным явлением, как и симптомы и признаки дефицита жирорастворимых витаминов; линия на диаграммах роста может показать снижение.
Диагностика
- Общий и прямой билирубин.
- Функциональные тесты печени.
- Гепатобилиарное сканирование.
- Иногда биопсия печени.
Любой младенец, который имеет желтуху в возрасте 2 нед, должен быть обследован на холестаз. Первоначальный подход должен быть направлен на диагностику состояний, поддающихся лечению (например, атрезии внепеченочных желчных протоков, при которой раннее хирургическое вмешательство улучшает исход).
Холестаз идентифицируют по увеличению как общего, так и прямого билирубина. Тесты, которые необходимы для дальнейшей оценки функции печени, включают определение альбумина, фракционированного билирубина в сыворотке крови, ферментов печени, протромбинового времени (ПВ) и АЧТВ. После подтверждения холестаза необходимо провести обследование для определения этиологии.
Должно быть проведено гепатобилиарное сканирование; экскреция контрастного вещества в кишечник исключает атрезию желчных протоков, но отсутствие экскреции может быть как при атрезии желчных протоков,так и при тяжелом неонатальном гепатите. Неонатальный гепатит характеризуется лобулярным расстройством с гигантскими многоядерными клетками. Иногда диагноз остается неясным, и необходимо проведение диагностического хирургического вмешательства с оперативной холангиографией.
Прогноз неонатального холестаза у детей
Прогноз холестаза из-за наличия специфических расстройств (например, метаболических заболеваний) варьируется начиная от доброкачественного течения до прогрессирующего заболевания, приводящего к циррозу печени.
Синдром идиопатического неонатального гепатита обычно проходит медленно, но в результате может развиться необратимое повреждение печени, приводящее к смерти.
Лечение неонатального холестаза у детей
- Лечение специфических причин.
- Пищевые добавки витаминов A, D, Е и К.
- Среднецепочечные триглицериды.
- Иногда урсодезоксихолевая кислота.
Специфическое лечение направлено на причину. Если специфическая терапия отсутствует, лечение поддерживающее и состоит в основном из лечебного питания, в т.ч. добавок витаминов А, D, Е и К. При искусственном вскармливании нужно использовать смеси, богатые среднецепочечными триглицеридами, поскольку они лучше всасываются в случае дефицита желчных солей. Требуется адекватное количество калорий; младенцам может потребоваться >130 калорий/кг в сутки. У новорожденных со сниженным желчеотделением применение урсодезоксихолевой кислоты может облегчить зуд
Младенцы с предполагаемой атрезией желчных путей требуют диагностической операции с интраоперационной холангиограммой. При подтверждении атрезии желчных путей должна быть проведена портоэнтеростомия (методика Касаи). В идеале эта процедура должна быть выполнена в первые 1-2 мес жизни. После операции многие пациенты имеют значительные хронические проблемы, в т.ч. стойкий холестаз, рецидивирующий восходящий холангит и задержку в развитии. Даже при оптимальной терапии у многих младенцев развивается цирроз, и им требуется трансплантация печени.
Читайте также:
