
Молекулярная модель вируса гриппа
В нашем первом посте про трехмерное моделирование вирусов мы перечислили основные стадии процесса и рассказали о том, с чего мы начинаем и как собираем исходную информацию. В этой заметке мы расскажем о следующем этапе работы — о создании моделей отдельных молекул, из которых впоследствии будет собрана целая частица.

Компоненты вирусной частицы Гриппа A/H1N1
Вирусная частица — это молекулярный механизм, решающий две принципиальные задачи. Во-первых, частица должна обеспечить упаковку вирусного генома и его защиту от деструктивных факторов среды, пока вирус путешествует из клетки, в которой он собрался, к клетке, которую он сможет заразить. Во-вторых, частица должна быть способна присоединиться к заражаемой клетке, после чего доставить вирусный геном и сопутствующие молекулы внутрь, чтобы запустить новый цикл размножения. Задач не очень много, поэтому вирусы, за редким исключением, могут позволить себе быть довольно экономными в том, что касается структуры.
В частности, геном большинства вирусов невелик и кодирует не очень много белков, нередко это число меньше 10. При этом вирус может заставить клетку синтезировать большое количество однотипных белков, из которых потом соберется вирусная оболочка — капсид. Таким образом, вирусные частицы обычно состоят из большого числа одинаковых элементов, которые связываются друг с другом как детали конструктора, часто образуя регулярные и симметричные структуры. Так, очень многие, хоть и не все вирусные упаковки или их фрагменты имеют спиральную или икосаэдрическую форму.

Примеры вирусных капсидов с икосаэдрической симметрией. Молекула бактриородопсина в правом нижнем углу — для сравнения. (Иллюстрация из обзора).
Для сборки модели вируса принципиально важно знать, как устроены отдельные белки общей структуры и как они друг с другом связываются, эту структуру формируя. Современная наука владеет целым набором методов, которые могут дать ответы на эти вопросы, однако ни один из подходов, к сожалению, не является универсальным и решает только часть задач которые стоят перед нами при создании научно достоверных моделей вирусов с атомной детализацией.
Напомним, что белки — это полимерные молекулы, состоящие из последоватльно связанных между собой мономеров — аминокислот. В водных растворах белки обычно сворачиваются в сложные трехмерные глобулы (почти как головоломка “Змейка Рубика”), форма которых зависит от аминокислотного состава и некоторых других факторов. Пространственное строение этих глобул определяют в основном методами рентгеноструктурного анализа и ЯМР-спектроскопии. Также в последнее время к этой задаче позволяет подойти электронная микроскопия.
В целом, методы определения пространственной структуры молекул сложны и имеют целый набор ограничений, поэтому далеко не все вирусные белки описаны полностью. Так, рентгеноструктурный анализ предполагает наличие кристалла, через который пропускается рентгеновское излучение. Атомы кристалла провоцируют дифракцию рентгеновских лучей, по картине которой можно оценить распределение электронных плотностей в кристалле, а по этим данным уже восстановить расположения конкретных атомов. Этот метод дает разрешение вплоть до чуть более 1 ангстрема (0,1 нм), однако в случае белков проблема заключается в том, что далеко не все из них можно кристаллизовать. Особенно сложным это оказывается, если белок имеет гибкие подвижные или заякоренные в мембране фрагменты.
ЯМР-спектроскопия основана на явлении ядерного магнитного резонанса и позволяет описывать строение белков в растворе. Этот подход выявляет набор возможных положений атомов в молекуле и, в отличие от предыдущего метода, дает возможность оценить степень гибкости тех или иных ее участков. Но ЯМР-спектроскопия хорошо работает только для сравнительно небольших молекул, поскольку крупные белки дают слишком много шума.
Электронная микроскопия позволяет описать строение крупных молекулярных комплексов, что бывает очень полезно, когда речь идет о вирусах. Для многих симметричных структур можно получить большой набор изображений под разными углами, проанализировав которые можно воссоздать трехмерную картину. Для отдельных объектов разрешение, получаемое в результате применения разных вариантов электронной микроскопии (до 4-5 ангстрем), оказывается не многим хуже разрешения рентгеноструктурного анализа, хотя обычно для получения полной информации приходится совмещать разные подходы и, например, “вписывать” структуры отдельных белков в карты электронных плотностей, получаемые при помощи электронной микроскопии.

Структуры тримера белка оболочки ВИЧ (красные и голубые фрагменты молекул) в комплексе с участком одного из антител к этому белку (зеленые и желтые фрагменты), вписанные в карту электронной плотности, полученную методом крио-электронной микроскопии с разрешением 9 ангстрем. Из статьи Structural Mechanism of Trimeric HIV-1 Envelope Glycoprotein Activation.
Как мы писали в прошлом посте, получаемые структуры систематизируются и хранятся в базе данных Protein Data Bank. При этом в формате *.pdb записываются координаты атомов, и существует целый набор программ, позволяющих эти данные визуализировать и работать с такими структурами. Среди них, например VMD, Chimera, PyMol и десятки других.

Скриншот текстового отображания файла в формате *.pdb. Описываются координаты отдельных атомов в аминокислотах белка.
Программы могут отображать белки несколькими способами. Помимо простого отображения атомов сферами разного диаметра, соответствующего ван-дер-ваальсовым радиусам атомов, существует возможность показать отдельные связи, поверхность молекулы, а также изгибы аминокислотной цепочки при помощи структур, напоминающих ленты (ribbon diagram), которые наглядно демонстрируют, где в белке аминокислоты образуют альфа-спирали, где бета-слои, а где неструктурированные участки.

Различные варианты визуализации структуры наружней части гемагглютинина вируса гриппа в программе Chimera.
В качестве отступления, надо сказать, что программы, в которых обычно работают ученые, визуализируя отдельные молекулы или белковые комплексы, чаще всего позволяют получить лишь довольно примитивные с эстетической точки зрения результаты (достаточно, например, посмотреть на несколько скриншотов из программы VMD). Принципиально более широкие возможности открываются, если импортировать модели молекул в программы, которые используют профессиональные дизайнеры и специалисты компьютерной трехмерной графики. Эти программы в сочетании с плагинами, улучшающими качество рендера, позволяют получать действительно интересные и привлекательные визуализации. Мы еще расскажем об этом в следующих постах. Пока просто приведем пример:
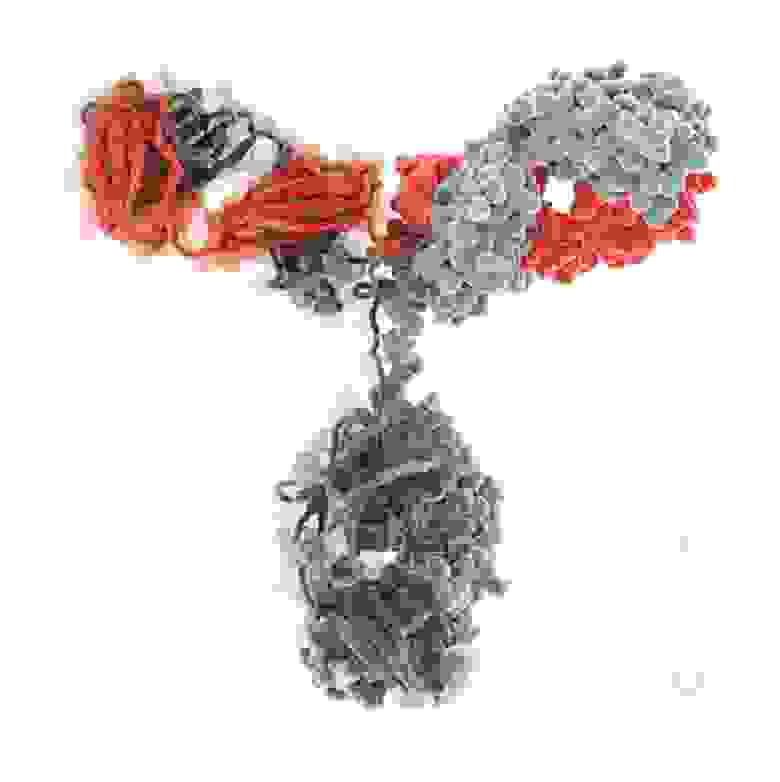

Изображения молекулы иммуноглобулина G.

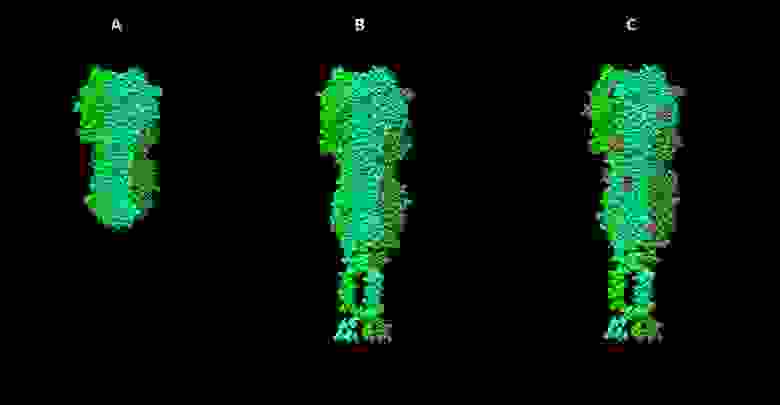
Шаблоны для моделирования нейраминидазного комплекса вируса гриппа. А — фрагмент мономера нейраминидазы N2 из структуры 2AEP в базе данных PDB, B — “стебель” гемагглютинин-нейраминидазы парагриппа (3TSI), С — трансмембранный пептид 2LAT. D — финальная полученная модель.
Окончательная модель белка обычно создается с учетом известных структур его фрагментов, найденных разными методами шаблонов, а также моделей от сервера I-Tasser. Для этого используется программа Modeller. Она позволяет строить модель по гомологии с использованием одного или нескольких шаблонов, а также вносить дополнительные модификации, например, создавать дисульфидные связи в заданных местах.
Другим важным аспектом строения вирусов, информация о котором в научной литературе часто оказывается не полна, является взаимодействие между отдельными белками. В нашем случае от этого зависит то, какими поверхностями модели отдельных белков будут контактировать друг с другом и другими компонентами вириона в финальной модели. Информацию о взаимодействиях тоже позволяет уточнить структурная биоинформатика.
Программа докинга не моделирует естественный процесс образования комплекса, это было бы слишком медленно и ресурсоемко, а перебирает варианты взаимного положения двух или более молекул в поисках наилучшей структуры. При докинге обычно большую молекулу в комплексе называют рецептором, а меньшую — лигандом. Для определения качества структуры комплекса лиганда с рецептором используются различные оценочные функции. В идеале в качестве такой функции должна выступать свободная энергия системы, но она слишком сложно вычисляется, поэтому применяют различные эмпирические псевдопотенциалы, учитывающие потенциальную энергию (которая как раз вычисляется просто), площадь контакта лиганда и рецептора, соответствие различным правилам, которые исследователи вывели из анализа большого числа комплексов, и всякие загадочные слагаемые, не имеющие физического смысла, но улучшающие результат программы при испытании на большом количестве известных комплексов. Поиск минимума такого псевдопотенциала в современных программах обычно происходит с помощью различных вариаций метода Монте-Карло и генетических алгоритмов. В настоящее время существует множество программ молекулярного докинга (наиболее известные из них — Dock, Autodock, GOLD, Flexx, Glide), отличающиеся оценочными функциями, методами минимизации и дополнительными возможностями. При этом во время поиска молекулы рецептора и лиганда могут как оставаться неподвижными (такой тип докинга называется жестким), так и несколько менять конформацию (гибкий докинг). Очевидно, что второй вариант более ресурсоемкий, но и результаты такого поиска обычно правдоподобнее. Докинг малых молекул к белкам сейчас является стандартным этапом разработки новых лекарственных препаратов. Можно, например, провести докинги для 10 миллионов лигандов, и выбрать сотню наиболее перспективных соединений для дальнейшей экспериментальной работы — это называется виртуальный скрининг.
Помимо исследований небольших молекул, докинг может быть использован и для построения белок-белковых и белок-нуклеотидных комплексов. Для этих целей также разработано большое количество программ и онлайн-сервисов (ZDOCK, pyDOCK, HEX). Например, в ходе нашей работы над вирусом папилломы человека (ВПЧ) мы столкнулись с тем, что, несмотря на наличие полной структуры внешнего слоя капсида, образованнного белком L1, совершенно не было информации о строении белка L2, который в капсиде расположен ближе к геному, а соответственно, нет данных о том, как пентамеры L1 взаимодействуют с молекулами L2. Мы построили модель белка L2 по гомологии, используя сервер Tasser, после чего провели докинг в программе HeX. В ходе докинга роль рецептора выполнял пентамер L1. Именно на его поверхности проводился поиск оптимального места посадки L2. При этом все структуры оставались неподвижными. Т.е. использовался метод жесткого докинга. В результате была получена правдоподобная структура комплекса пентамера, собранного из L1 и минорного белка L2.
Наконец, биоинформатическими методами можно пытаться восстановить то, какие изменения в структуру вирусных белков вносит сама клетка, в которой они образуются. Большинство белков после синтеза подвергаются дополнительным химическим посттрансляционным модификациям (ПТМ), которые могут серьезно влиять на выполняемые белком функции. Среди таких модификаций фосфорилирование, убиквитинирование, гликозилирование, нитрозилирование, внесние разрывов и другие химические изменения. Многие поверхностные белки вирусов гликозилированы, причем эта модификация имеет непосредственное значение для выполнения основной функции поверхностных белков вируса — связывания с клеточными рецепторами. С другой стороны, белки вирусных матриксов — слоев, которые встречаются непосредственно под липидными оболочками некоторых вирусов, для заякоривания в мембране часто должны быть связаны, например, с миристиловой кислотой — небольшой гидрофобной молекулой, облегчающей взаимодействие белков с липидами. Таким образом, в нашей работе модификации белков тоже требуют внимания.
В настоящее время возможные ПТМ достаточно сложно предсказываются. Основные существующие методы и сервисы основаны на поиске соответствующей экспериментальной информации для сходных белков или поиске в последовательности исследуемого белка небольших участков, характерных для того или иного типа модификации.
В нашей работе при подготовке моделей мы пользуемся экспериментальной информацией, отраженной в соответствующей записи базы данных UNIPROT.
Стадии работы над моделью гемагглютинина вируса гриппа. А — визуализация структуры 3ZTJ из базы данных PDB. B — модель гемагглютинина вируса гриппа H1N1, построенная на основе гомологии с 3ZTJ с достраиванием трансмембранных участков молекулы. С — модель с учетом посттрансляционных модификаций (гликозилирования).
Последнее, о чем хочется упомянуть, — это то, что при подготовке новых моделей белков и, особенно, их комплексов, необходимо проводить оптимизацию структур. Наиболее простым методом оптимизации является минимизация энергии. Она используется для достаточно быстрого “спуска” системы в локальный минимум потенциальной энергии. Эту манипуляцию желательно проводить после каждой модификации структуры молекул. Она позволяет избежать таких неприятностей, как перекрывание атомов или появление неправильных длин связей. Различные методы минимизации энергии предусмотрены практически в любом программном пакете молекулярного моделирования.
Стоит отметить, что данный метод позволяет провести лишь предварительную и очень грубую оптимизацию. Для более точной подготовки пространственных структур используются методы молекулярной динамики или квантовой механики. Последние, например, используются для наилучшей оптимизации структуры небольших молекул лигандов и наиболее точных расчетов энергии межмолекулярных взаимодействий. Но, наибольшая точность, что вполне логично, связана с более ресурсоемкими вычислениями, что делает эти методы практически неподъемными в применении к большим биологическим макромолекулам.
Оценить поведение и стабильность структур достаточно массивных молекул, таких как полипептиды и нуклеиновые кислоты позволяют методы молекулярной динамики.
Метод молекулярной динамики заключается в изучении поведения атомов и молекул и их движений во времени. Расчеты молекулярной динамики позволяют, например, исследовать стабильность как отдельных молекул, так и их комплексов, позволяют оценить значимость возможных конформационных перестроек, влияние точечных мутаций и многое другое. Современные методы анализа результатов симуляций молекулярной динамики позволяют получить самые подробные сведения о поведении во времени как отдельных атомов, так и всей исследуемой системы.
В зависимости от того, насколько хорошо изучены белки того вируса, модель которого мы хотим создать, каждый раз приходится подбирать подходы для достройки и оптимизации моделей всех белков и их взаимодействий. После того, как все структуры получены, можно приступать к сборке полной модели. О том, как это делается, мы расскажем в следующих постах серии о создании научно достверных моделей вирусов человека.
PS:
Ставшая лидером в опросе прошлого поста тема Медицинская анатомическая иллюстрация — история изучения тела человека в работах иллюстраторов 5 столетий будет следующей. С потрясающими гравюрами, восковым моделями прошлого века, пластификатами трупов, атласами выдающся исследователей, 3Д реконструкциями на основе послойных срезов замороженного смертника, интерактивными приложениями и работами современных медицинских иллюстраторов. Скоро.
Вирус гриппа — это широко распространенный, легко передающийся и быстро эволюционирующий возбудитель заболевания. Симптомы гриппа могут напоминать признаки обычной простуды, однако болезнь чревата осложнениями, которые особенно опасны для маленьких детей, пожилых людей и тех, кто страдает хроническими заболеваниями. Новые штаммы гриппа возникают ежегодно. Штаммы — это разновидности вируса, отличающиеся друг от друга двумя поверхностными белками, которые быстро накапливают изменения и тем самым увеличивают их разнообразие. В случае одновременного заражения организма разными штаммами белки могут попадать в формирующиеся в этом процессе новые частицы в произвольных сочетаниях, что вновь ведет к росту разнообразия вирусов. Это, в свою очередь, вызывает дополнительные проблемы с лечением инфекции и увеличивает риск межвидового заражения.
Грипп встречается не только у людей, но и среди животных. Циркулируя в их популяциях, вирус нередко приобретает наиболее неприятные для человека свойства.
История эпидемий гриппа
Успехи в разработке противовирусных препаратов и профилактических мер, а также активный мониторинг распространения и возникновения новых штаммов гриппа позволяют сокращать число жертв и интенсивность эпидемий. Однако и сейчас в результате вызываемых им осложнений погибает от 250 до 500 тысяч человек в год.
Международный проект компании Visual Science"Зоопарк вирусов" — первая успешная попытка создать модели наиболее распространенных и опасных вирусов человека с разрешением до атомов.
Задача построения научно достоверной 3D-модели вируса не так тривиальна, как кажется на первый взгляд. Ни один из имеющихся на сегодняшний день научных подходов, будучи применён по отдельности, не позволяет получить изображение целой вирусной частицы в атомном или даже молекулярном разрешении. На молекулярном уровне вирусы представляют собой структуры огромного размера: один вирус — это комплекс сотен макромолекул (белков,нуклеиновых кислот, в ряде случаев липидов). Несмотря на это, вирусы слишком малы для того, чтобы их можно было досконально изучить, используя электронную и тем более оптическую микроскопию: эти инструментальные методы хорошо подходят для исследования строения существенно более крупных биологических объектов, например клеток. В частности, электронная микроскопия даёт возможность получить грубые изображения, на которых видны лишь контурывириона (этот метод не всегда позволяет увидеть даже крупные поверхностные белки).
Для изучения отдельных белков широко используются методы рентгеноструктурной кристаллографии и ядерного магнитного резонанса. Полученные с их помощью данные представляют собой пространственные координаты атомов, входящих в состав молекулы, в наиболее энергетически выгодном положении из возможных. В контексте задачи визуализации структуры вириона недостатки этого метода связаны с невозможностью исследования с его помощью более массивных объектов (он неприменим даже к небольшим комплексам нескольких белков) и трудностью изучения подвижных и связанных с мембранами компонентов вируса.
Таким образом, задача создания научно достоверных 3D-моделей напоминает сборку паззла, где в исходном наборе не хватает значительной части фрагментов, отдельные фрагменты являются неполными, а иллюстрация результата, к которому стремятся собирающие паззл исследователи, груба, и потому даёт только общее представление о конечной картине. Тем не менее, сотни работ разных авторов со всего мира проливают свет на многие вопросы структуры и морфологии компонентов вирионов, а также их взаимодействия. При тщательном анализе всех научных работ, с учетом мнений признанных экспертов из мировых научных центров и помощи специалистов Отдела молекулярного моделирования компании Visual Science, устраняющих пробелы в имеющихся исследованиях по молекулярной биологии, вирусологии и кристаллографии, появляется возможность создать максимально точные и достоверные модели вирусов, которые и представлены в проекте Viral Park.
Компоненты вируса: более 200 тысяч молекул 11 типов
Вирион гриппа имеет форму удлинённой по одной из осей сферы диаметром 80-120 нанометров, что в тысячи раз меньше толщины человеческого волоса. Продолговатую форму вируса определяет слой структурного белка — матрикса. С внешней стороны матрикс окружён мембраной с поверхностными белками (гемагглютинином и нейраминидазой) и ионными каналами. Геном вируса — это восемь молекул РНК в составе спиральных комплексов. Он, как и белки, необходимые для полноценной работы вируса в зараженной клетке, находится внутри частицы.
— Гемагглютинин связывается с рецепторами на поверхности клетки, позволяя частице слиться с мембраной клеточной везикулы, проникнуть внутрь нее и доставить геном вируса в цитоплазму.
— Нейраминидаза нужна для того, чтобы только что сформированные вирионы могли отделяться от клеточной мембраны и заражать другие клетки.
— Белок матрикса — одна из основных структурных молекул вируса. Именно с этим белком связана характерная форма вирусной частицы и расположение в ней внутренних компонентов (что, в свою очередь, детерминирует правильность распаковки и процесса заражения), он же определяет её размеры. Молекулы белка матрикса связываются как с поверхностными белками, так и с нуклеопротеиновыми комплексами вируса, позволяя всем компонентам попадать в частицу.
— Белок M2 образует каналы, через которые внутрь частицы проникают ионы водорода после того, как та оказывается в клетке. Это запускает разборку вируса и высвобождение его генома.
— Нуклеопротеин упаковывает фрагменты вирусного генома в компактные спирально закрученные комплексы геномной РНК и белка NP — рибонуклеопротеиды (РНП),которые помещаются внутри вирусной частицы.
— Белок ядерного экспорта обеспечивает транспорт копий РНК генома из ядра, где они образовались, к месту сборки новых частиц вируса у поверхности зараженной клетки.
— Полимеразный комплекс необходим для создания копий РНК вируса, одна часть которых нужна для того, чтобы синтезировать структурные белки, а другая — чтобы упаковываться в новые вирусные частицы. Геном вируса несет информацию, необходимую для синтеза вирусных белков. Он представлен восемью молекулами РНК, отличающимися длиной и набором кодируемых белков.
— Мембрана вируса формируется из мембраны клетки, в которой он образовался. В ее состав входят молекулы фосфатидилхолина, фосфатидилэтаноламина, сфингомиелина и холестерина — в характерных для клеток человека пропорциях.
Процесс создания научно достоверных 3Д моделей вирусов
Пространственные структуры некоторого количества белков, входящих в состав вирионов (этадоля варьируется от 25 до 50%), описаны не полностью: проблемы возникают из-за того, что не все белковые молекулы или отдельные их фрагменты возможно кристаллизовать (а это является условием проведения рентгеноструктурного анализа). Обычно в структурах недостаёт подвижных фрагментов молекул, а также трансмембранных участков и гликозильных остатков поверхностных белков. В то же время для комплексов и ансамблей белков в большинстве случаев неизвестно, какие поверхности компонентов формируют контакты друг с другом, а какие — нет. Предсказание и описание таких взаимодействий необходимо при построении любой вируснойчастицы (для большей части входящих в неё молекулярных комплексов и структур).
Эти проблемы позволяет решить структурная биоинформатика, основанная на вычислительных методах: в данном подходе используется предсказание пространственной структуры исследуемой молекулы на основе структур схожих или родственных протеинов и исходной последовательности аминокислот, которая в большинстве случаев известна заранее. Кроме того, подобные методы позволяют рассчитать меж- и внутримолекулярные взаимодействия. В компании Visual Science эту задачу решает Отдел молекулярного моделирования и динамики: работающие в нем специалисты — молекулярные биологи и биоинформатики, обладающие научными степенями. Они проводят анализ опубликованных ранее кристаллографических данных (параллельно оценивая точность описания похожих белков), осуществляют моделирование недостающих фрагментов и в результате проводят сборку полных достоверных моделей всех компонентов и комплексов вирусной частицы, комбинируя известные данные с теми, что получены ими в Отделе. Подобный уровень сотрудничества с научным сообществом, а также столь высокая степень вовлечения в этот процесс структурных биологов из числа сотрудников Отдела молекулярного моделирования компании на данный момент недоступны ни одной студии научной и медицинской визуализации в мире — кроме Visual Science.
Дополнительные материалы: научный обзор и панорама 3Д модели вируса гриппа.

Меньше не имело смысла
В поисках информации консультанты Visual Science пишут письма вирусологам по всему миру и время от времени находят неопубликованные данные
В отличие от черных дыр, вирусы повсюду. С ними умеют работать ученые, им посвящены тысячи теоретических и экспериментальных работ, однако их внешний вид все равно остается загадкой: ученым-вирусологам очень редко нужно знать, как выглядит вирус. Его взаимодействие с клетками человека или бактериями не включает в себя визуального контакта. Оптические микроскопы не могут показать объект, линейные размеры которого уступают длине световой волны: фотографии вирусов выглядят так, как будто у камеры сбился фокус. И даже эти размытые изображения из университетских учебников двухмерные.
Создать красивую и достоверную модель – это просветительский проект. Стефанов, который отвечает за достоверность моделей, признает, что решает не научную задачу. Впрочем, количество научных статей, которые ему приходится перелопатить во время работы над следующим вирусом, не дает соскучиться по профессии. Несколько лет назад, уже защитив диссертацию, он ушел в Visual Science из академического института.

Трудная жизнь
Вирус – самая простая живая структура, но невероятно сложная по меркам неживого мира. Он включает в себя до нескольких десятков белков, каждый из которых состоит из десятков-сотен тысяч атомов и имеет сложную пространственную структуру. Генетическая информация в виде ДНК или РНК намотана на эти белки часто неизвестным науке образом. Работа над новым объектом начинается с составления карты белых пятен, неизученных деталей строения вируса.
В парке вирусов, который надеется создать Visual Science, есть сравнительно простые и трудные объекты. Сложность работы определяется в первую очередь тем, насколько хорошо изучен вирус и какие белые пятна исследователям приходится закрывать собственными усилиями. Пространственная структура многих белков неизвестна.
Взаимодействие молекул иногда приходится выяснять самостоятельно, методами биоинформатики. В поисках информации консультанты Visual Science пишут письма вирусологам по всему миру и время от времени находят неопубликованные данные, которые можно пустить в ход. Главный успех компании – вирус гриппа – состоялся благодаря помощи испанских биологов, которые как раз готовили статью в Science, посвященную укладке его генетического материала. В результате сотрудничества вирус появился на обложке.
Поскольку вирусы меньше длины световой волны, у них нет цвета. Сотрудники Visiual Science исправляют этот эстетический недостаток, а заодно добавляют в модель дополнительную информацию. Белки, которые кодируются собственным геномом вируса, раскрашиваются цветом, а белки, которые кодируются зараженной клеткой, изображаются в черно-белой гамме (вирусы заставляют механизмы человеческой клетки работать на воспроизводство своих поработителей). Когда какой-то пурист в интернете указал Константинову на это очевидное погрешение против истины, он специально обесцветил модель вируса, чтобы продемонстрировать, как скучно тот выглядит без цвета.
Физические размеры вируса, как ни странно, тоже усложняют моделирование: с ростом числа молекул геометрически растет необходимое для расчетов компьютерное время. Самые большие известные вирусы поражают, на счастье, амеб, а не людей, поэтому не интересуют Visual Science. Но даже на рендеринг вируса Эбола, заметно более крупного, чем грипп или ВИЧ, коллеги потратили месяц компьютерного времени. Неделю потеряли из-за того, что во дворе дома, где проводились расчеты, под землю провалился КамАЗ и отрубил питание в лаборатории.
Человеческих вирусов хватит еще на много лет работы. Например вирус гепатита, при его упоминании у Стефанова появляется огонек в глазах. Когда-нибудь можно будет замахнуться на кусок настоящей живой клетки, но это потом: они на несколько порядков больше вирусов.
Вы работали в Институте молекулярной генетики РАН, и именно там к вам пришла идея создать Visual Science? Что послужило толчком?
Идея пришла раньше, когда я учился на биологическом факультете МГУ на кафедре молекулярной биологии. Мы изучали такие дисциплины, как молекулярная биология, биохимия, анатомия — то есть то, что связано с объяснением устройства сложных пространственных объектов. Даже в лучших западных учебниках удивлял достаточно низкий визуальный и методический уровень материалов. И в то же время мы видели кино и игры с совершенно потрясающей 3D-графикой, которая используется исключительно для увеселения и развлечений. Так возникла идея совместить технологии кино-, игровой и рекламной индустрий с научной экспертизой для эффектной подачи наукоемких понятий и данных без потери достоверности.
Далеко не каждый ученый может стать предпринимателем. Были ли какие-то сомнения или решение было однозначным?
Предпринимательство — это один из самых эффективных путей личного развития. Кроме того, это сейчас можно зарабатывать в науке достойные деньги, а 10 лет назад все было по-другому, так что и меркантильный фактор при создании компании тоже присутствовал. В этом смысле предпринимательство мне показалось оптимальным вариантом.
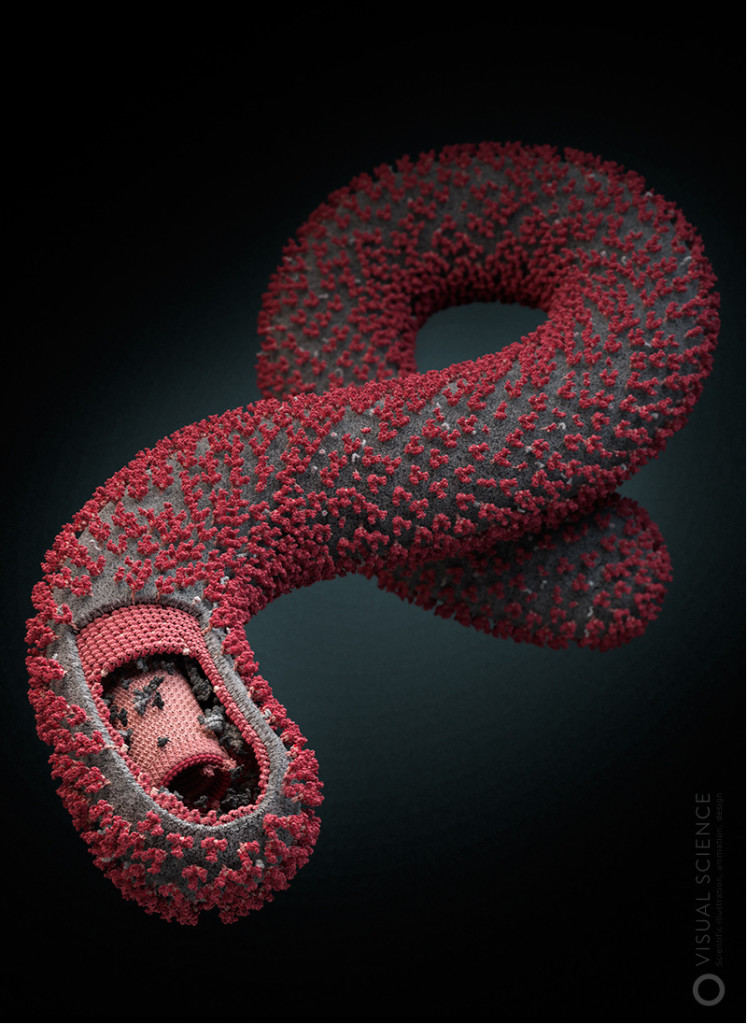
Модель вируса Эбола.
Визитная карточка Visual Science — это моделирование вирусов. А как вообще можно смоделировать то, чего не видно?
Уточню, что в целом нашей визитной карточкой является hi-end научная визуализация в самых разных сферах. От молекулярной биологии и нанотехнологий до медицины, ядерной физики, высокотехнологичных производств и так далее. Возвращаясь к вашему вопросу, скажу, что как раз то, чего не видно, и нужно моделировать. Научное моделирование, которым мы занимаемся, состоит в том, что мы собираем данные сотен научных публикаций и исследований ведущих ученых, дополняем результатами моделирования нашего научного отдела и совмещаем их воедино в модели с высокой степенью достоверности и детализации, которая в то же время красива, интересна как специалистам, так и широкому кругу.
Откуда у моделей вирусов берется цвет? Разве это не влияет на достоверность модели?
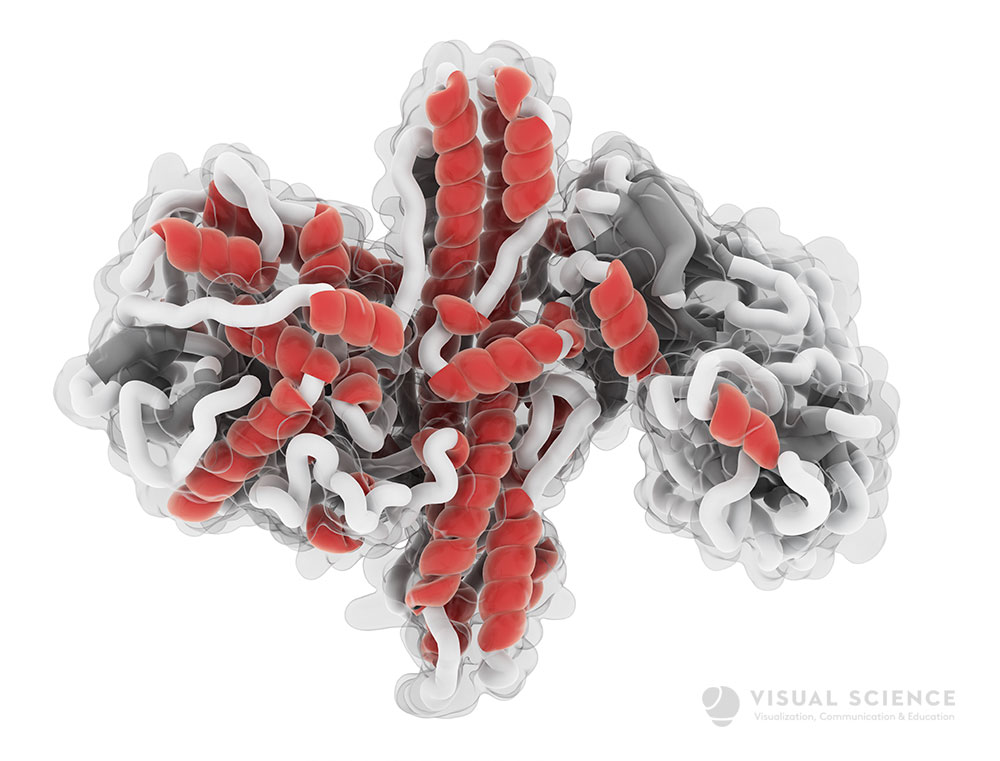
Модель ботулотоксина A, одного из самых опасных ядов на Земле. В медицинских целях его используют для разглаживания морщин.
Проект очень интересный, но как частная компания вы должны зарабатывать деньги. Кто ваш типичный клиент и что является конечным продуктом для покупателя?
У нас 4 основных направления. Во-первых, это hi-end научная визуализация и анимация. В рамках этого направления мы помогаем представить сложные концепции и объекты в наглядной и понятной форме. Это интересно как нобелевским лауреатам, для которых мы создаем графику для лекций, так и био- и нанотехнологическим, фармацевтическим компаниям, университетам и даже глянцевым журналам. Практически все издательства научной литературы в мире, а также издательства научно-популярных журналов являются нашими клиентами.

Второе направление — дизайн и коммуникация. Это решение полного цикла дизайнерских и коммуникационных задач для фармацевтических, медицинских, бионанотехнологических компаний. Нашими клиентами являются различные компании, от небольших перспективных стартапов до представителей ТОП-10 крупнейших фармацевтических компаний мира.
Третье направление — образование. От пластиковых моделей и классических учебников до интерактивных курсов с дополненной и виртуальной реальностью — все, что нужно для преподавания точных наукоемких дисциплин в наше время.
Четвертое — образовательные пособия и музейные экспонаты. Здесь нашими клиентами являются Дарвиновский, Политехнический музеи и другие естественнонаучные музеи, которые создают современные экспозиции и выставки. Также мы консультируем компании, которые курируют создание новых музеев в США, и предоставляем им контент.
Как Visual Science устроена изнутри?
У нас есть вычислительный центр, который позволяет рассчитывать ресурсоемкие проекты. Например, вирус Эбола состоит из нескольких миллионов частиц, и это очень ресурсоемкая задача. Коллектив компании состоит из ряда специалистов, которые живут и работают по всему миру, и большого пула внештатных научных советников. Это ученые из ведущих исследовательских центров, от Гарварда до Гонконгского университета науки и технологий. Они помогают нам обеспечить экспертизу на самом высоком уровне. Помимо этого, в Москве расположена лаборатория образовательных пособий и музейных экспонатов. Это небольшое производство, на котором мы создаем прототипы и музейные экспонаты малыми сериями.
Сколько стоит создать модель вируса? Какие есть границы по деньгам от самой дешевой визуализации до самой дорогой?
Если говорить об иллюстрациях, от нескольких сотен до 40–50 тысяч долларов за изображение. Все зависит от сложности объекта, глубины экспертизы и необходимого уровня детализации. Над некоторыми проектами мы работаем год и более.
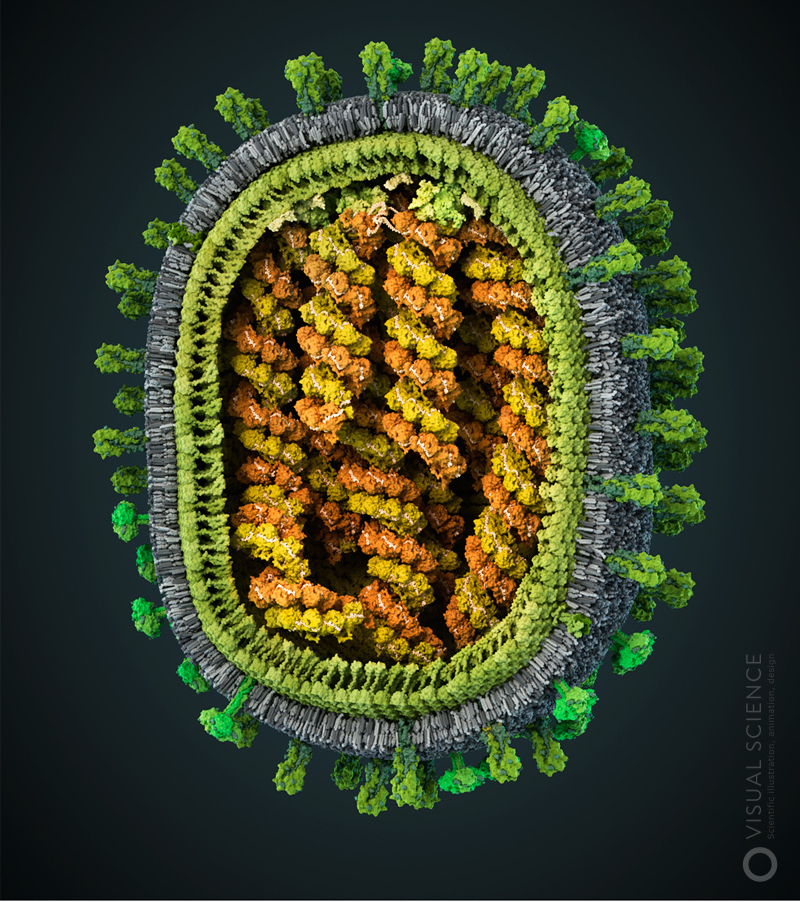
Модель вируса гриппа.
На главной странице вашего сайта сейчас красуется модель вируса гриппа. Это предмет особой гордости вашей компании. А как создавалась эта модель?
Модель вируса папилломы человека.
Используете ли вы технику Apple для создания моделей и если да, то какую?
Для повседневной работы самыми удобными оказались MacBook серии Pro в максимальной конфигурации. К сожалению, приходится держать виртуальные машины, так как значительная часть научного софта сделана под Windows. Вычислительные станции работают на Windows. Что касается научного моделирования, тут практически все сделано под Linux.
Сейчас, находясь в 2015 году, верили ли вы в 2007 году, в момент открытия компании, что все сложится именно так? Каким вы видите будущее Visual Science?
Верил — да, знал — нет. Все меняется очень быстро, мы сделали несколько пивотов в процессе развития. Мы начинали как компания, которая занималась только биомедицинской визуализацией. Это оказалось слишком узкой нишей. Клиенты хотели большего. До декабря 2014 года мы налаживали мелкосерийное производство кастомных образовательных пособий, но скачок курса доллара заставил нас изменить планы, подход стал нерентабелен. К пособиям скоро вернемся, правда, на других принципах. А вообще будущее компании я связываю с образованием, популяризацией точного и научно достоверного подхода к познанию мира, налаживанием коммуникации научного сообщества с широкой аудиторией. Речь идет прежде всего о естественных науках, которые требуют точности, наглядного и понятного объяснения.
Спасибо и удачи вам в развитии компании.
Читайте также:
