
Гепатит с при фенилкетонурии
Фенилкетонурия – это наследственное заболевание, связанное с нарушением метаболизма аминокислот, в частности, фенилаланина. Это вещество вместе с его токсическими продуктами накапливается в организме ребенка и приводит к тяжелому поражению центральной нервной системы. Больные фенилкетонурией страдают от нарушения умственного развития.

Патология чаще наблюдается у девочек, чем у мальчиков. Если родители здоровы, но являются гетерозиготными носителями мутантного гена, их дети в большинстве случаев наследуют фенилкетонурию. Также заболевание присуще малышам, родившимся в результате родственных браков. По статистике фенилкетонурия у детей встречается чаще всего в странах Европы, где на 100000 новорожденных один имеет признаки отклонения, а также в Ирландии и в России. В африканских странах болезнь практически отсутствует.
У больных фенилкетонурией наблюдается нехватка фермента фенилаланин-4-гидроксилаза, обеспечивающего превращение фенилаланина в тирозин, что, собственно, и является сущностью заболевания. Накопление вещества и его производных нарушает ЦНС, белковый обмен, метаболизм гормонов, обмен липо- и гликопротеидов. Также происходит расстройство транспорта аминокислот, сбои в обмене катехоламинов и серотонина.
Симптомы фенилкетонурии
Несмотря на то, что фенилкетонурия наследуется, первые признаки можно заметить не с самого рождения. Только с 3-6 месяца родители замечают у малыша вялость, повышенную раздражительность, отсутствие интереса к окружающим предметам, рвоту, беспокойство.
Симптомы фенилкетонурии во втором полугодии выражаются отставанием в психическом развитии. Около 10% малышей страдают слабой степенью олигофрении, 60% — идиотией. При фенилкетонурии у детей рост может быть как и нормальным, так и недостаточным. Зубки у таких малышей прорезываются, как правило, позже, чем у здоровых, также наблюдается уменьшение черепа. Больные фенилкетонурией начинают гораздо позже сидеть и ходить. Таких людей можно узнать визуально по своеобразной позе и походке: в положении стоя ноги широко расставлены и согнуты в коленных и тазобедренных суставах, при этом голова и плечи опущены. Ходят же больные фенилкетонурией мелкими шажками, слегка покачиваясь. Из-за повышенного мышечного тонуса они вынуждены сидеть, поджав ноги.
Чаще всего симптомом фенилкетонурии является белесость кожного покрова, который полностью лишен пигмента. Волосы у больных чаще светлые, глаза – голубые. Нередко окружающие замечают своеобразный запах от страдающих фенилкетонурией, который напоминает мышиный. Также у некоторых больных наблюдаются эпилептические припадки, которые с возрастом проходят.
К симптомам фенилкетонурии относят повышенную потливость, синюшность конечностей и дермографизм, из-за чего человек становится более подвержен негативному влиянию солнца и травм. У детей, наследовавших фенилкетонурию, часто встречается тяжелая экзема, дерматит, склонность к запорам и артериальная гипотония.

Диагностика фенилкетонурии
Прогноз заболевания напрямую зависит от своевременности постановки диагноза и соответствующего лечения. Несмотря на то, что симптомы патологии появляются не раньше третьего месяца жизни, наличие отклонений можно определить уже сразу после рождения. Таким образом, если малыш родился доношенным, то на 4-5-й день у него берут кровь на анализ на данное заболевание. Если же малыш родился недоношенным, тогда диагностику проводят на 1-7 день. При выявлении концентрации фенилаланина более 2,2 мг, родителей и ребенка направляют на дополнительное обследование в медико-генетический центр.
Лечение фенилкетонурии
На данный момент единственным лечением фенилкетонурии является диетотерапия. С раннего возраста малыш может питаться материнским молоком, но при этом матери необходимо соблюдать рекомендации врача. Специальная диета для лечения фенилкетонурии полностью исключает пищу, содержащую белок. В рационе не должно быть мяса, орехов, рыбы, яиц, творога, бобовых, круп, хлебобулочных изделий, шоколада и других продуктов. Жизненно важные элементы больные получают из специальных смесей, которые заменяют белок. Жир человек может получать из растительного и сливочного масла. Предпочтение отдается фруктам, сокам, овощам. Страдающие фенилкетонурией должны находиться под регулярным контролем психоневролога и педиатра.
При обнаружении фенилкетонурии с первых дней жизни анализ крови проводится каждую неделю до момента нормализации показателей. После этого необходимо сдавать этот же анализ каждый месяц в течение первого года жизни ребенка. У детей постарше диагностика проводится один раз в несколько месяцев.
Фенилкетонурия (ФКУ) — это наиболее распространенное врожденное нарушение метаболизма аминокислот, возникающее в результате дефицита фермента фенилаланингидроксилазы. Вследствие данного заболевания снижается способность организма к метаболизму аминокислоты фенилаланина. Эти процессы вызывают накопление фенилаланина в тканях организма и физиологических жидкостях.
Повышенные уровни фенилаланина негативно влияют на когнитивные функции, поэтому у людей с классической фенилкетонурией почти всегда присутствует интеллектуальная инвалидность. Заболевание доходит до этой стадии, если уровни фенилаланина не контролируются путем диетической или медикаментозной терапии.
Механизм развития и причины фенилкетонурии
Механизм, вследствие которого повышенные уровни фенилаланина вызывают интеллектуальную инвалидность, в точности не известен, однако ограничение продуктов, содержащих вредные вещества, снижает этот эффект.
В детском возрасте уровень фенилаланина в крови и коэффициент интеллекта (IQ) тесно связаны.
В настоящее время в ведущих клиниках мира ведется детальное изучение незначительных нейропсихологических дефицитов у детей с контролируемой фенилкетонурией. Некоторые ученые списывают этот дефицит на остаточные аномалии функции нейромедиаторов — например, снижение производства нейротрансмиттеров в результате недостаточного транспорта тирозина сквозь нейронную клеточную мембрану.
Иногда уровни аминокислот у больных в норме, но присутствует дефицит синтеза или утилизации белка BH4. Такое состояние называется злокачественной фенилкетонурией. BH4 кофактор необходим для гидроксилирования тирозина (предшественника дофамина) и триптофана (предшественника серотонина). Таким образом, пациенты с дефицитом BH4 могут иметь дополнительные проблемы неврологического характера, которые невозможно полностью скорректировать диетическим ограничением количества фенилаланина. Таким пациентам необходимы дополнительные методы лечения, которые не гарантируют стопроцентной эффективности.
Фенилкетонурия — это аутосомно-рецессивное заболевание, вызванное мутацией в гене ФАГ (фенилаланингидроксилазы). На сегодняшний день учеными идентифицировано более 500 различных мутаций гена ФАГ.
Для фенилкетонурии характерна заметная генотипическая гетерогенность, как внутри определенной популяции, так и между различными популяционными группами населения. Генотип и фенотип людей с данным заболеванием тесно связаны, кроме того, неродственные индивидуумы с одинаковыми мутациями имеют определенную степень изменчивости толерантности к фенилаланину. Генотип — это совокупность наследственных генетических характеристик, а фенотип — это совокупность внешних факторов среды, влияющая на человека уже после рождения.
ФКУ является наследственным заболеванием, вызванным дефектом в гене ФАГ. Ген ФАГ помогает создать фенилаланингидроксилазу — фермент, ответственный за разрушение фенилаланина. Опасное накопление фенилаланина может произойти, если человек употребляет чрезмерное количество продуктов с высоким содержанием белка, например, яйца и мясо. Оба родителя должны иметь дефектную версию гена ФАГ, только тогда ребенок наследует расстройство. Если только один родитель является носителем измененного гена, ребенок не будет иметь каких-либо симптомов, но так же будет являться носителем гена.
Диагностика фенилкетонурии
С 1960-х годов в больницах Соединенных Штатов был введен регулярный скрининг новорожденных на ФКУ путем взятия пробы крови. В настоящее время такая диагностика всё еще актуальна. Проводится взятие пробы крови из пятки новорожденного, а затем анализируется на ФКУ и другие генетические нарушения.
Скрининг-тест выполняется, когда новорожденному исполняется один-два дня, и он еще находится в роддоме.
Дополнительные тесты могут быть выполнены, чтобы подтвердить первоначальные результаты. Обычно врачи проверяют ген ФАГ на наличие мутаций, которые вызывают фенилкетонурию.
Если ребенок или взрослый проявляет симптомы фенилкетонурии, например, очевидна задержка развития, необходимы дополнительные анализы крови для подтверждения диагноза. Тест подразумевает взятие образца крови и ее анализ на присутствие фермента, необходимого для расщепления фенилаланина.
Симптомы фенилкетонурии
Большинство детей с фенилкетонурией кажутся нормальными при рождении. Если анализ крови после рождения не был проведен, а заболевание осталось невыявленным, происходит прогрессивная задержка развития — наиболее распространенный симптом фенилкетонурии.
Первые симптомы фенилкетонурии у новорожденных:
У пожилых людей с фенилкетонурией на МРТ отчетливо видны проявления демиелинизации. Присутствует заторможенность когнитивных функций и двигательных навыков. Коэффициент умственного развития снижается на 10 единиц и более, если прекратить придерживаться рекомендуемой врачом диеты.
- изменение цвета волос и оттенка кожи, являющееся следствием ухудшения синтеза меланина.
Больной имеет волосы светло-рыжего или белого цвета и болезненно бледную кожу, голубые глаза, светлые ресницы и брови.
Такое изменение цвета волос и кожи может быть характерным даже для афроамериканцев и азиатов, однако это скорее редкость, чем норма.
Другие симптомы кожных проявлений фенилкетонурии:
- экзема (атопический дерматит);
- увеличение числа гнойных инфекций;
- фолликулярный кератоз;
- потеря волос.
Симптомы фенилкетонурии могут быть как легкими, так и интенсивными. Наиболее тяжелой формой расстройства является так называемая классическая фенилкетонурия. Ребенок с классическим типом заболевания может казаться абсолютно нормальным первые несколько месяцев жизни. Если заболевание не выявляется и не лечится, появляются первые симптомы, которые затем усиливаются.
К таким симптомам относятся:
Менее тяжелая форма фенилкетонурии — это состояние, при котором у новорожденного в организме слишком большое количество фенилаланина. Дети с этой формой заболевания могут иметь только легкие симптомы, но они должны придерживаться специальной диеты, чтобы избежать развития умственных нарушений.
Если фенилкетонурия не диагностируется при рождении и лечение не начинается быстро, расстройство может вызвать:
- необратимое повреждение мозга и психические расстройства в течение первых нескольких месяцев жизни;
- поведенческие проблемы и судороги у детей более старшего возраста.
Лечение фенилкетонурии
Пациенты с фенилкетонурией могут облегчить тяжесть симптомов и предотвратить развитие осложнений, следуя специальной диете и принимая лекарства.
Самой эффективной диетой является ограничение продуктов, содержащих фенилаланин. Дети с ФКУ не могут потреблять грудное молоко, они питаются по специальной схеме, употребляя продукт Лофеналак или его аналоги — продукты, специально предназначенные для людей с таким заболеванием.
Если ребенок достаточно взрослый, чтобы жевать твердую пищу, необходимо полностью исключить такие продукты:
Для того чтобы больные могли получать достаточное количество белка, дети с ФКУ употребляют специальные пищевые добавки, содержащие все аминокислоты, необходимые организму, за исключением фенилаланина.
Существуют также определенные продукты с низким содержанием белка, которые можно найти в специализированных магазинах или аптеках. Эти пищевые добавки больные должны употреблять всю жизнь, чтобы избежать снижения умственных способностей и появления различных неприятных симптомов.
Все пациентам с фенилкетонурией необходимо постоянно контактировать с диетологом или лечащим врачом, чтобы правильно поддерживать надлежащий баланс питательных веществ, ограничивая при этом потребление фенилаланина. Также необходимо контролировать уровни фенилаланина путем ведения учета количества фенилаланина в продуктах питания, которые человек потребляет в течение дня.
Управление качеством пищевых продуктов и медикаментов США (FDA) недавно одобрило препарат Сапроптерин (Kuvan) для лечения фенилкетонурии. Sapropterin помогает снизить уровень фенилаланина. Этот препарат должен быть использован в сочетании со специальной схемой употребления пищи. Однако наиболее эффективен он для людей с легкими формами фенилкетонурии.
Беременность и фенилкетонурия
Женщина с фенилкетонурией в период беременность подвергается риску осложнений, если не следует схеме питания, рекомендованной врачом. Среди возможных рисков следует отметить вероятность выкидыша. Также есть шанс, что пока еще нерожденный ребенок будет подвергаться воздействию высоких уровней фенилаланина. Это может привести к различным проблемам у плода, в том числе:
- ограниченным интеллектуальным возможностям;
- порокам сердца;
- задержке роста;
- низкому весу на момент рождения;
- аномально маленькой голове (микроцефалии).
Все эти признаки становятся заметными у новорожденного не сразу, но тест, проведенный в первые сутки – двое после рождения, может помочь определить наличие заболевания.
Иногда лечение больных с фенилкетонурией (ФКУ) проводят в специальных клиниках, профилем которых являются метаболические нарушения. Обычно схема лечения разрабатывается комплексно, при участии эндокринолога, иммунолога и диетолога.
Уровни фенилаланина следует проверять через регулярные промежутки времени, примерно 1-2 раза в неделю у новорожденных и один раз в месяц для детей старшего возраста и взрослых.
Большинство американских клиник рекомендуют удерживать уровень фенилаланина в пределах 2-6 мг/дл (120-360 мкмоль/л). Точное попадание в рекомендуемый диапазон показателей требует квалифицированного ухода и тщательного мониторинга.
Диету ни в коем случае не рекомендуется прекращать по достижении подросткового возраста, поскольку гиперфенилаланинемия может иметь пагубные последствия и для взрослых пациентов.
Некоторые взрослые пациенты с незарегистрированной фенилкетонурией и, следовательно, интеллектуальной инвалидностью, могут значительно улучшить когнитивные показатели и физическое состояние при соблюдении специальной диеты.
Больные, проходящие качественное лечение, могут придерживаться нормального уровня активности.
Прогноз при фенилкетонурии
Прогноз для разных групп пациентов сильно отличается. Например, дети, заболевание которых было обнаружено в раннем возрасте (в течение первого месяца жизни), при тщательном контроле состояния здоровья, соблюдении режима питания, могут жить абсолютно нормальной жизнью.
Если ребенок не придерживается диеты или соблюдает её частично, нарушения умственного развития неизбежны.
Большинство больных с фенилкетонурией, не имеющих никакого лечения, принадлежат к категории людей с глубокой интеллектуальной инвалидностью.
Распространенные трудности таких больных это:
- психологические проблемы, например, агорафобия и другие фобии;
- нарушение внимания;
- пониженная способность к концентрации;
- нарушения поведения.
Долгосрочный прогноз для пациентов с ФКУ очень хороший, если сразу после рождения ребенка родители придерживаются специальной диеты. Интеллектуальная инвалидность без лечения наступает уже в первый год жизни.
Проявляется она в следующем:
- заторможенность;
- нарушения поведения;
- неспособность к адекватному выражению эмоций (ребенок не узнает родителей, капризничает и т.д.);
- неврологические проблемы (судороги, тремор).
Можно ли предотвратить фенилкетонурию?
ФКУ является генетическим заболеванием, поэтому она не может быть предотвращена. Однако, ферментный анализ — это один из способов узнать, являются ли муж или жена носителем дефектного гена.
Всем больным с ФКУ следует избегать аспартама (искусственного подсластителя). Аспартам широко используется в производстве лекарственных средств, витаминов, напитков и других съедобных продуктов.
Диета с исключением фенилаланина увеличивает вероятность метаболических проблем, например, снижает способности организма к усвоению фолиевой кислоты или жирных кислот.
Как в любом возрасте быстро отучиться грызть ногти?
Часто задаваемые вопросы
Фенилкетонурия (ФКУ) – довольно редкое наследственное заболевание, связанное с нарушением обмена аминокислот. Организм больного фенилкетонурией человека не способен расщеплять аминокислоту фенилаланин, которая поступает с белковой пищей. В результате этого, в тканях накапливаются соединения, отравляющие нервную систему и головной мозг в частности. Развивается умственная отсталость (малоумие), вплоть до идиотии. В связи с этим болезнь получила и другое название – фенилпировиноградная олигофрения.
Однако из всех наследственных заболеваний фенилкетонурия, единственное, которое удается полностью нейтрализовать. Сегодня ребенка, рожденного с признаками ФКУ, можно вырастить абсолютно здоровым. Обезопасить мозг малыша удается с помощью специальной диеты, о которой мы расскажем ниже.
В разных странах частота этого заболевания отличается в разы. В России рождается один больной ребенок на 10 000. В некоторых регионах Великобритании этот показатель в два раза выше – 1:5000. Дети на Африканском континенте практически не болеют фенилкетонурией. Среди больных количество девочек почти в два раза превышает количество мальчиков.
Механизм развития заболевания
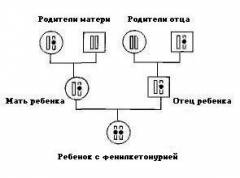
Заболевание наследуется только в том случае, если оба родителя передали ребенку склонность к болезни, и поэтому встречается довольно редко. У двух процентов людей есть измененный ген, который отвечает за развитие болезни. При этом человек остается полностью здоровым. Но когда мужчина и женщина, носители мутировавшего гена, вступают в брак и решают завести детей, то вероятность того, что малыши будут страдать от фенилкетонурии, составляет 25%. А возможность того, что дети будут носителями патологического гена ФКУ, но сами останутся практически здоровыми, составляет 50%.
Причина возникновения этого заболевания связана с тем, что в печени человека не вырабатывается особый фермент – фенилаланин-4-гидроксилаза. Он отвечает за превращение фенилаланина в тирозин. Последний входит в состав пигмента меланина, ферментов, гормонов и необходим для нормальной работы организма.
При ФКУ фенилаланин, в результате побочных путей обмена, превращается в вещества, которых не должно быть в организме: фенилпировиноградную и фенилмолочную кислоты, фенилэтиламин и ортофенилацетат. Эти соединения накапливаются в крови и оказывают комплексное действие:
- нарушают процессы жирового обмена в мозге
- вызывают дефицит нейромедиаторов, которые передают нервный импульс между клетками нервной системы
- оказывают токсическое действие, отравляя мозг
Симптомы фенилкетонурии

Дети с ФКУ рождаются абсолютно здоровыми. Поэтому, если в течение первых дней жизни выявить заболевание и придерживаться диеты, то удается предотвратить разрушение мозга ребенка. При этом, никакие признаки заболевания не появляются. Малыш развивается и растет, как и его сверстники.
Если же момент упущен, и ребенок употребляет в пищу белковые продукты, богатые фенилаланином, то начинают проявляться симптомы поражения центральной нервной системы. Поначалу изменения у больных фенилкетонурией незначительны. Их трудно заметить даже опытному педиатру. Это слабость и беспокойство. Малыш не улыбается и мало двигается.
Годовалый ребенок не умеет выражать голосом свои эмоции и переживания, имеет невыразительную мимику, не понимает речь родителей.
В возрасте трех лет и старше симптомы фенилкетонурии нарастают. У детей наблюдается повышенная возбудимость, утомляемость, нарушения поведения, психотические расстройства, умственная отсталость. Если не заниматься лечением фенилкетонурии, то состояние больного будет ухудшаться.
Диагностика фенилкетонурии

В том случае, если есть подозрение, что один или оба родителя являются носителями гена ФКУ, то определить это можно в федеральных медико-генетических центрах. Для установления этого факта проводится генетическая экспертиза.
На сегодняшний день все новорожденные дети массово обследуются на наличие фенилкетонурии. На территории России этот вопрос регламентирует приказ Минздрава РФ №316 от 30.12.1993 г. Процедура получила название неонатальный скрининг и является эффективным способом выявления наиболее распространенных наследственных заболеваний, среди них и ФКУ.
Массовое обследование новорождённых это простой и достоверный метод диагностики. В роддоме у каждого ребенка берут несколько капель периферической крови из пяточки. Это делается натощак, через три часа после кормления. У доношенных детей анализ берут на четвертый день жизни, а у недоношенных на седьмой. У тех новорожденных, которые появились на свет не в родильных домах, важно взять анализ на протяжении первых трех недель.
В том случае, если в анализе обнаруживают измененный ген, то родителей с ребенком приглашают в медико-генетический центр для обследования. Для того, чтобы подтвердить или опровергнуть диагноз назначаются дополнительные исследования:
- в сухом пятне крови
- в сыворотке крови
- потовый тест
- копрограмма
- ДНК-диагностика
Лечение фенилкетонурии
На сегодняшний день в нашей стране единственным эффективным методом лечения является диетотерапия. Разрабатываются препараты, которые позволят контролировать уровень фенилаланина в крови без соблюдения диеты. В этом направлении есть значительные успехи, но в продаже такие лекарственные средства появятся не раньше чем через 5-7 лет.
Постоянно идет работа над поиском новых средств и методов борьбы с болезнью.
- Перспективным направлением считается использование растительного фермента фенилаланинлиазы, который будет расщеплять излишки фенилаланина в организме.
- Ученые возлагают большие надежды на генотерапию с использованием вирусного фактора, которая позволит вылечить больной ген и полностью избавиться от проблемы.
- Практикуется введение гена фенилаланингидроксилазы прямо в пораженные клетки печени.
Некоторые формы фенилкетонурии поддаются лечению тетрагидробиоптерином, который является составляющей частью недостающего фермента фенилаланин-4-гидроксилазы. Атипичные формы ФКУ не лечатся с помощью диеты и требуют регулярного приема тетерагидробиоптерина или его заменителей.
Питание больного фенилкетонурией

Для того, чтобы нервные клетки ребенка не подвергались токсическому воздействию фенилаланина и его производных, нужно полностью исключить из рациона животные белки. Если это сделать на первых неделях жизни, то мозг останется полностью здоровым. Если же начинать ограничивать белок в более позднем возрасте, то задержку развития удается несколько приостановить. Но вернуть здоровье нервной системе и устранить изменения в нервных клетках уже не удастся.
Соблюдать диету требуется до 16-18 лет. Это обязательное условие. Желательно контролировать количество животных белков и в дальнейшем.
Если женщина, у которой в детстве были обнаружены признаки ФКУ, планирует забеременеть, то ей обязательно нужно вернуться к диете без фенилаланина. Таких ограничений необходимо придерживаться до зачатия, во время беременности и кормления грудью.
Все необходимые для роста и развития аминокислоты поступают в организм из специализированных лечебных продуктов. Обычно они представляют собой порошок – сухую смесь аминокислот. Их родителям больного малыша выдают бесплатно в медико-генетической консультации.
Грудные дети получают специальные смеси полностью очищенные от лактозы на основе гидролизата молочного белка.
Детей, больных ФКУ, можно кормить грудью. Но при этом кормящей матери нужно придерживаться специальной диеты.
В диете детей дошкольного и школьного возраста полностью исключают из меню белковые продукты. В списке разрешенных продуктов – овощи, фрукты, изделия из крахмала, растительные масла. При составлении дневного меню необходимо строго придерживаться возрастных норм фенилаланина.
| Возраст ребенка | Суточное количество фенилаланина (мк/кг массы тела) |
| Младше 2 мес. | 60 |
| 2-3 мес. | 60-55 |
| 3-6 мес. | 55-45 |
| 6-12 мес. | 45-35 |
| 1-1,5 года | 35-30 |
| 1,5-3 года | 30-25 |
| 3-6 лет | 25-15 |
| Старше 6 лет | 15-10 |
Необходимо помнить, что для растущего организма полноценное питание жизненно необходимо. Так в сутки ребенку требуется 120 мг тирозина на каждый килограмм массы. Поэтому дети и подростки с таким диагнозом должны получать аминокислоты для построения клеток и роста из дополнительных источников. Также обязательно назначают витаминно-минеральный комплекс. Особенно важно, чтобы ребенок получал норму витаминов С, В6 и B1, фолиевой кислоты, железа, кальция и магния. Количество калорий должно быть увеличено на 30% по сравнению с дневной нормой сверстников.
Группы продуктов при ФКУ
Разделяют три группы натуральных продуктов. В основе классификации лежит количество в них фенилаланина:
- Красный список – продукты, которые необходимо полностью исключить из рациона.
- Оранжевый список – разрешены в небольших количествах под строгим контролем.
- Зеленый список – могут употребляться без ограничений.
| Красный список | Оранжевый список | Зеленый список |
| Все виды мяса | Молочные продукты | Фрукты |
| Колбасные изделия | Рис и кукуруза | Ягоды |
| Все виды рыбы | Овощи (картофель, капуста) | Зелень |
| Морепродукты | Овощные консервы | Овощи |
| Яйца | Рисовая, кукурузная мука | |
| Сыры | Крахмал и саго | |
| Творог | Сахар и варенье | |
| Орехи | Мед | |
| Хлеб и хлебобулочные изделия | Сливочное и растительное масло, топленый жир | |
| Кондитерские изделия | ||
| Крупы и хлопья | ||
| Продукты из сои | ||
| Поп-корн | ||
| Аспартам |
Промышленность выпускает еще две группы продуктов:
- искусственные низкобелковые продукты, специально для диетического питания (хлеб, печенье, макароны)
- готовые пюре для детского питания на основе фруктов.
Родителям ребенка, больного ФКУ, важно уметь составлять диету и правильно рассчитывать количество фенилаланина. Для этого необходимо иметь под рукой весы, которые дают возможность взвешивать до десятой доли грамма.
Контроль уровня фенилаланина в крови

Необходимо контролировать количество фенилаланина. Оно должно находиться в границах 3–4 мг% или 180–240 мкмоль/л.
Для определения необходимо сделать анализ крови в лаборатории. До трехмесячного возраста это делают еженедельно.
Постепенно врач снижает количество анализов. С тех месяцев до года – один раз в месяц, с года до трех лет – один раз в два месяца. После трех лет частота проверок снижается до одного раза в три месяца. Существует специальная схема, но специалист может изменить ее, исходя из состояния больного.
Делать анализ желательно утром натощак. От качества и регулярности такого контроля и своевременных исправлений в диете зависит сохранение интеллекта.
Ответы на часто задаваемые вопросы
Новорожденные с диагнозом ФКУ ничем не отличаются от здоровых детей. И если болезнь вовремя выявить и остановить ее развитие, то и в дальнейшем такой ребенок останется абсолютно здоровым.
Как выглядят больные фенилкетонурией? 
Для того чтобы обеспечить детей всеми необходимыми веществами в достаточном количестве разработаны специальные смеси на основе незаменимых и заменимых аминокислот. В их состав также входят витамины и все необходимые микроэлементы.
Для детей до одного года рекомендуют:
- Афенилак 13, Афенилак 15 от компании "Нутритек", Россия;
- MIDмил ФКУ 0 (Hero, Испания);
- ХР Аналог ("Нутриция", Голландия);
- Фенил Фри 1 ("Мид Джонсон" США).
- П-АМ 1, П-АМ 2, П-АМ 3;
- Изифен (готовый продукт), а также ХР Максамейд и ХР Максамум с нейтральным и фруктовым вкусами ("Нутриция", Голландия).

На сегодняшний день в России для лечения ФКУ используют специальную безфенилаланиновую диету. Для пополнения запасов тирозина и других аминокислот, все больные дети бесплатно получают специальные препараты. Соблюдать диету желательно до 18 лет, хотя ряд врачей утверждает, что лучше делать это на протяжении всей жизни.
Подводя итоги, отметим, что фенилкетонурия это тяжелое генетическое заболевание, которое может привести к психической инвалидности. Изменения происходят в нервной системе очень быстро и имеют необратимый характер. Однако можно не допустить развития болезни. Для этого необходима ранняя диагностика и специальная диета.
- ФенилкетонурияI. Классическая и наиболее распространенная форма заболевания, описанная выше в статье. Связана с мутацией гена в 12-й хромосоме, при этом нарушается образование фермента фенилаланин-4-гидроксилазы, который превращает фенилаланин в тирозин.
- ФенилкетонурияII. При этой форме заболевания нарушение происходит в 4-й хромосоме. Нарушается выработка фермента дигидроптеридинредуктазы, который также способствует превращению фенилаланина в тирозин. Заболевание наследуется так же, как и I форма: для того, чтобы родился больной ребенок, необходимо, чтобы носителями гена были оба родителя. Распространенность фенилкетонурии II – 1 случай на 100 000 новорожденных.
- ФенилкетонурияIII. В результате генетических нарушений возникает недостаток фермента 6-пирувоилтетрагидроптеринсинтазы. Наследуется, как и две предыдущие формы заболевания. Распространенность – 1 случай на 300 000 новорожденных.
Критерии установления инвалидности при фенилкетонурии:
- При фенилкетонурии I инвалидность устанавливают только при необратимых нарушениях со стороны центральной нервной системы, которые приводят к неврологическим расстройствам и умственной отсталости.
- При фенилкетонурии II и III типа группу инвалидности устанавливают во всех случаях.

Специальной профилактики фенилкетонурии не существует. Но некоторые мероприятия помогают правильно оценить риски, вовремя принять необходимые меры:
- Генетическое консультирование. Необходимо людям, планирующим завести ребенка, которые больны или являются носителями неправильного гена, у которых болен хотя бы один близкий родственник или уже родился больной ребенок. Консультирование проводит врач-генетик. Он помогает разобраться, как ген, ответственный за фенилкетонурию, передавался в предыдущих поколениях, каковы риски будущего ребенка. Также генетик помогает с планированием семьи.
- Скрининг новорожденных. Анализ не помогает предотвратить заболевание, но позволяет выявить его максимально рано, пока оно еще не привело к необратимым изменениям в головном мозге.
- Консультации и диета для женщин, страдающих фенилкетонурией. Если вы женщина и страдаете ФКУ, вам следует проконсультироваться с врачом и спросить, когда лучше планировать беременность в вашем случае. Во время беременности нужно соблюдать правильную диету – это помогает предотвратить дефекты развития у ребенка.
Прогноз зависит от формы заболевания и начала лечения, соблюдения диетических рекомендаций, медико-педагогической коррекции.
При фенилкетонурии I, при своевременном начале необходимых мероприятий, прогноз, как правило, благоприятный. Ребенок растет и развивается нормально. Если опоздать с лечением и диетой, то результат будет не таким хорошим.
При фенилкетонурии II и III прогноз серьезнее. Соблюдение диеты не приносит эффекта.
Читайте также:
