
Сколиоз эхинококкоз синдром клайнфельтера

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Клайнфельтера, 47, XXY является клиническим примером поражения половых хромосом.
Болезнь Клайнфельтера характеризуются наличием как минимум одной лишней X хромосомой у мальчиков, что приводит к нарушению полового созревания у них. Клинически впервые описан Клайнфельтером в 1942 г. Популяционная частота составляет 1 : 1000 лиц мужского пола. Синдром Клайнфельтера встречается примерно у 1/800 родившихся живыми мальчиков. Лишнюю Х-хромосому ребенок получает от матери в 60 % случаев.

[1], [2], [3]
Код по МКБ-10
Что вызывает болезнь Клайнфельтера?
В большинстве случаев неправильное расхождение половых хромосом происходит в гаметах родителей. Имеют место и мозаичные варианты, например 47, XXY/46, XY.
Синдром Клайнфельтера обусловлен хромосомной патологией, представленной в наиболее типичном варианте как 47ХХУ. Значительно реже встречаются мозаичные формы - 46ХУ/47ХХУ. Как казуистические варианты кариотипа описаны формы 48ХХХУ, 47ХХУ/46ХХ, 47ХХУ/45ХО. Имеется также наблюдение пациента с кариотипом 47ХХУУ46ХХ/45ХО. Причиной данных хромосомных аномалий - дополнительной Х-хромосомы в мужском кариотипе - может являться нерасхождение Х-хромосомы в период первого или второго мейотического деления или нарушение митотического расхождения хромосом в период развития зиготы (мозаичные варианты). Метод ДНК-анализа позволил выявить, что 53% больных с синдромом Клайнфельтера имели дополнительную хромосому отцовского происхождения, являвшуюся следствием нерасхождения в период первого мейотического деления. 43% больных имели дополнительную хромосому материнского происхождения как результат патологии первого и второго мейотического деления. По-видимому, не имеется различий фенотипа у пациентов, имеющих дополнительную материнскую или отцовскую Х-хромосому Частота рождения мальчиков с синдромом Клайнфельтера увеличивается по мере увеличения возраста матери. Подобной зависимости от возраста отца не выявлено. Наличие в мужском кариотипе дополнительной Х-хромосомы не влияет на дифференцировку тестикул и формирование гениталий по мужскому типу. Однако жизнедеятельность герминативных клеток нарушается, сперматогенез отсутствует. Причиной этого является активность дополнительной Х-хромосомы в герминативных клетках, имеющих в норме гаплоидный набор хромосом. Было показано, что в герминативных клетках яичника плода у девочек перед вступлением в мейоз происходит реактивация второй Х-хромосомы (в норме активирована только одна). У мальчиков с кариотипом ХХУ также сохранен пред мейотический процесс реактивации второй Х-хромосомы, однако процесс расхождения нарушен, и герминативная клетка может содержать две активные Х-хромосомы, что приводит к ее гибели уже в первые дни после реактивации Х-хромосомы. У взрослых мужчин с синдромом Клайнфельтера при анализе клеток спермы единичные сохранные герминативные клетки имели только нормальный гаплоидный хромосомный набор.
Патогенез синдрома Клайнфельтера
Наличие лишней X хромосомы приводит к аплазии эпителия тестикул, которые в дальнейшем гиалинизируются. Это приводит у взрослых больных к азооспермии и бесплодию.
[4], [5], [6], [7]
Симптомы синдрома Клайнфельтера
При рождении синдром Клайнфельтера клинически не проявляется. Клинических вариантов, касающихся как аномалий полового статуса, так и соматических нарушений при синдроме Клайнфельтера, описано достаточно много. Общей закономерности влияния кариотипа на фенотип не выявлено, но пациенты, имеющие мозаичный кариотип с нормальным мужским клоном 47ХХУ/46ХУ, имеют менее тяжелые нарушения.
Первые отчетливые фенотипические признаки заболевания появляются в пре- и пубертатном периодах онтогенеза. До пубертатного возраста у мальчиков могут выявляться крипторхизм (чаще двусторонний) и маленькие размеры полового члена. У 50% мальчиков отмечается умеренная задержка умственного развития, сопровождающаяся нарушениями поведения, трудностями контакта со сверстниками. Мальчики обычно имеют длину тела выше средневозрастных показателей. Характерны относительно длинные конечности, избыточное жироотложение по женскому типу (евнухоидное телосложение).

Поздно появляются вторичные половые признаки. Наиболее характерным симптомом синдрома Клайнфельтера является гипоплазия яичек и полового члена (гипогонадизм и гипогенитализм). У 50% больных в периоде полового созревания выявляется гинекомастия. Имеет место неглубокое снижение интеллекта, что сказывается на школьной успеваемости. Взрослые пациенты склонны к алкоголизму, наркомании, гомосексуализму и асоциальному поведению, особенно в условиях стресса.
Пубертат, как правило, начинается в обычном возрасте, однако часто оволосение на лице низкое. У таких детей наблюдается предрасположенность к нарушениям обучения, у многих отмечается сниженный вербальный интеллект, нарушены слуховое восприятие и обработка информации, а также навыки чтения. Клиническая вариабельность значительная, многие мальчики и мужчины с кариотипом 47, XXY имеют обычную внешность и нормальный интеллект.
В пубертатном возрасте вторичное оволосение появляется в обычные сроки, отмечается также увеличение полового члена. Однако объем тестикул увеличивается незначительно, не превышая, как правило, 8 мл; яички имеют плотную консистенцию. Пубертатная гинекомастия, часто достаточно ранняя, выявляется у 40-50% мальчиков В дальнейшем у этих пациентов повышается риск развития карциномы молочных желез. Костное созревание обычно соответствует возрасту к моменту инициации пубертата, однако позже дифференцировка костей скелета задерживается в связи с недостаточностью секреции тестостерона. Линейный рост конечностей продолжается до 18-20 лет, что приводит к формированию евнухоидных пропорций тела, конечный рост больных, как правило, выше роста родителей. Постпубертатная инволюция тестикул приводит к гипогонадизму и потере фертильности. При гистологическом исследовании выявляется гиалиноз семенных канальцев и отсутствие сперматогенеза. Количество клеток Лейдига может быть нормальным, однако с возрастом они подвергаются атрофии.
В 15 % случаев наблюдается мозаицизм. Эти мужчины могут иметь детей. У некоторых мужчин может быть 3,4 и даже 5 Х-хромосом вместе с одной Y-хромосомой. С увеличением числа Х-хромосом возрастает тяжесть умственной отсталости и пороков развития.
[8], [9]
Синдром Клайнфельтера - хромосомная патология, обусловленная наличием в мужском кариотипе одной или нескольких дополнительных женских половых хромосом. Синдром Клайнфельтера характеризуется первичным гипогонадизмом, маленькими размерами тестикул, бесплодием, гинекомастией, неглубоким снижением интеллекта. Решающая роль в диагностике синдрома Клайнфельтера принадлежит кариотипированию; также проводится анализ фенотипических признаков, определение полового хроматина, экскреции фолликулостимулирующего гормона с мочой, спермограмма и пр. Лечение при синдроме Клайнфельтера включает гормональную терапию, возможно - оперативную коррекцию гинекомастии, однако полное излечение синдрома невозможно.

Общие сведения
Синдром Клайнфельтера – дисомия или полисомия по женской половой хромосоме, при которой у лиц мужского пола имеется не менее двух Х-хромосом и одна Y-хромосома. Синдром Клайнфельтера встречается с частотой 1 случай на 850-1000 новорожденных мальчиков. Среди детей, страдающих олигофренией, распространенность синдрома Клайнфельтера составляет 1–2%. Синдром получил название по фамилии американского врача Гарри Клайнфельтера, впервые описавшего его в 1942 г. Кариотип таких больных с дополнительной Х-хромосомой был определен в 1959 г. Поскольку ведущим клиническим проявлением синдрома Клайнфельтера является первичный гипогонадизм, ведением таких пациентов занимаются специалисты в области эндокринологии и андрологии.
Причины синдрома Клайнфельтера
Как и в случае синдрома Дауна, хромосомная аберрация при синдроме Клайнфельтера связана с нерасхождением хромосом (в последнем случае – половых) в процессе мейоза либо нарушением деления зиготы. При этом значительно чаще (в 60%) мальчики с синдром Клайнфельтера получают лишнюю материнскую Х-хромосому, чем отцовскую.
Среди возможных причин подобного рода хромосомных аномалий называются вирусные инфекции, поздняя беременность, неполноценность регуляторных механизмов материнской и отцовской иммунной системы.
При наличии лишней X-хромосомы развивается аплазия эпителия яичек, их последующая гиалинизация и атрофия, что во взрослом возрасте сопровождается азооспермией и эндокринным бесплодием. Среди причин мужского бесплодия синдром Клайнфельтера составляет 10%, о чем всегда должны помнить специалисты в области репродуктивной медицины.
Наиболее частым цитогенетическим типом является полный вариант синдрома Клайнфельтера с кариотипом 47,ХХY. Реже встречается мозаицизм (46XY/47XXY; 46XX/47XXY), еще реже – полисомия 48,XXXY; 48,XXYY; 49,XXXXY и т. д. При мозаичном варианте (около 10% случаев) часть клеток имеет нормальный кариотип, поэтому мужчины с синдром Клайнфельтера могут иметь нормально развитые и функционирующие половые железы и сохранные репродуктивные способности.
Симптомы синдрома Клайнфельтера
Ребенок с синдромом Клайнфельтера рождается с нормальными росто-весовыми показателями, правильной дифференцировкой наружных гениталий, обычными размерами тестикул. В раннем возрасте у мальчиков с синдромом Клайнфельтера может отмечаться частая заболеваемость ОРВИ, бронхитом, пневмониями. Такие дети обычно отстают в моторном развитии (позднее начинают держать головку, сидеть, стоять, ходить), имеют задержку речевого развития. Уже в возрасте 5-8 лет мальчики с синдромом Клайнфельтера отличаются высоким ростом, диспропорциональным телосложением (длинными конечностями, высокой талией). В допубертатном возрасте может обнаруживаться одно или двусторонний крипторхизм.
Умственная отсталость умеренной степени, трудности установления контакта со сверстниками, нарушения поведения отмечаются у половины больных синдромом Клайнфельтера.
Отчетливые внешние признаки, свидетельствующие о наличии у ребенка синдрома Клайнфельтера, проявляются в препубертатном и пубертатном периодах развития. К ним относятся евнухоидный тип телосложения, позднее появление вторичных половых признаков, гипоплазия яичек, малый половой член, гинекомастия. В постпубертатном периоде онтогенеза наблюдается инволюция тестикул, сопровождающаяся потерей фертильности. При осмотре подростка с синдромом Клайнфельтера выявляется отсутствие или скудный рост волос на лице и в подмышечных впадинах, оволосение на лобке по женскому типу. У большинства больных присутствуют редкие поллюции, эрекция, сохранно половое влечение, однако из-за выраженного андрогенного дефицита в среднем к 30 годам происходит снижение либидо и развивается импотенция.
Синдрому Клайнфельтера часто сопутствуют аномалии скелета (деформации грудной клетки, остеопороз), нарушения прикуса, врожденные пороки сердца и др. Характерно преобладание ваготонических реакций: брадикардии, акроцианоза, потливости ладоней и стоп. Со стороны органа зрения нередко отмечается нистагм, астигматизм, птоз века.
Больные с синдромом Клайнфельтера предрасположены к развитию сопутствующих заболеваний: эпилепсии, рака молочной железы, сахарного диабета, ХОБЛ, желчнокаменной болезни, варикозного расширения вен, ожирения, гипертонической болезни, ИБС, ревматоидного артрита, острого миелоидного лейкоза. Могут отмечаться психические заболевания - маниакально-депрессивный психоз, шизофрения и др. Есть данные, подтверждающие склонность больных с синдромом Клайнфельтера к алкоголизму, наркомании и гомосексуализму.
Диагностика синдрома Клайнфельтера
Как и другие хромосомные аномалии, синдром Клайнфельтера у плода может быть обнаружен еще на этапе беременности при проведении инвазивной пренатальной диагностики (амниоцетеза, биопсии хориона или кордоцентеза с последующим анализом кариотипа или КФ-ПЦР).
Постнатальная диагностика синдрома Клайнфельтера проводится эндокринологами, андрологами и генетиками. При исследовании полового хроматина в клетках слизистой оболочки полости рта присутствуют тельца Бара, что является маркером синдрома Клайнфельтера. Другими характерными признаками служат особые изменения кожного рисунка на пальцах. Тем не менее, окончательный диагноз хромосомной аномалии может быть установлен только после исследования кариотипа.
УЗИ мошонки выявляет уменьшение объема яичек. При исследовании андрогенного профиля уровень тестостерона в крови больных синдромом Клайнфельтера понижен, однако при этом отмечается повышение уровня фолликулостимулирующего и лютеинизирующего гормонов. При анализе спермограммы выявляется олиго- или азооспермия. Морфологическое исследование материала, полученного путем биопсии яичек, выявляет гиалиноз семенных канальцев, гиперплазию клеток Лейдига, уменьшение числа клеток Сертоли, отсутствие сперматогенеза.
В течение жизни мужчины с синдромом Клайнфельтера могут обращаться к андрологу, сексологу, эндокринологу с проблемами бесплодия, импотенции, гинекомастии, остеопороза и др., однако нередко основное заболевание так и остается нераспознанным.
Лечение синдрома Клайнфельтера
Полностью излечиться от синдрома Клайнфельтера не представляется возможным. Тем не мене, все больные нуждаются в проведении симптоматической и патогенетической терапии. В детском возрасте необходима профилактика инфекционных заболеваний, закаливание, занятия ЛФК, коррекция нарушений речи с помощью логопеда.
С подросткового возраста пациентам с синдромом Клайнфельтера назначается пожизненная заместительная терапия половыми гормонами (внутримышечные инъекции тестостерон-пропионата, сустанона-250; сублингвальный прием метилтестостерона и др.). Ранняя и адекватная гормонотерапия препятствует атрофии яичек, способствует повышению полового влечения, развитию вторичных половых признаков. При резко выраженном увеличении молочных желез проводится операция по коррекции гинекомастии.
С целью повышения трудоспособности и социальной адаптации, предупреждения психопатизации личности и ее асоциальной направленности показана психотерапия.
Прогноз и профилактика синдрома Клайнфельтера
Пациенты с синдромом Клайнфельтера имеют нормальную продолжительность жизни, однако склонность к развитию хронических заболеваний может стать риск-фактором ранней смертности. Большинство больных с синдромом Клайнфельтера бесплодны; единственно возможным вариантом рождения детей в семьях, где партнер болен, является использование донорской спермы. Тем не менее, при мозаичной форме синдрома Клайнфельтера мужчины могут стать отцами самостоятельно или воспользовавшись вспомогательными репродуктивными технологиями (ЭКО).
Для оценки вероятности рождения ребенка с синдромом Клайнфельтера в процессе ведения беременности женщинам предлагается прохождение пренатального скрининга. Однако даже в случае получения положительных данных за наличие синдрома Клайнфельтера у плода настаивание на прерывании беременности со стороны акушера-гинеколога является недопустимым. Решение вопроса о целесообразности пролонгирования беременности должно приниматься родителями. При нормальном кариотипе родителей риск повторного появления ребенка с такой же хромосомной аномалией составляет не более 1%.
Диспансерное наблюдение больных с синдромом Клайнфельтера осуществляется эндокринологом.
Эхинококк – это гельминт, представитель группы цестод, рода лентиформных червей, относящийся к отряду циклофиллид, половозрелая особь которого чаще всего обитает в полости тонкой кишки собак, волков, кошек.
Опасным для человека является заражение личинками паразита – эхинококкоз.
При этой нозологии в различных органах (лёгкие, печень, желудок, мышцы) происходит образование однокамерных или многокамерных кист с персистирующими видами.
Долгое время к эхинококкозам ошибочно приписывали схожее поражение альвеококком, потому что это такое же глистное заболевание, со схожими рентгенологическими данными и стертой клинической картиной. Для верификации двух паразитов необходима биопсия.
Тело половозрелой особи червя (стробила) состоит из 3-7 члеников, а общая длина варьируется от 2 до 7 мм. На головке имеется прикрепительный аппарат, состоящий из 4 присосок и двойного венчика из 30-40 крючьев.
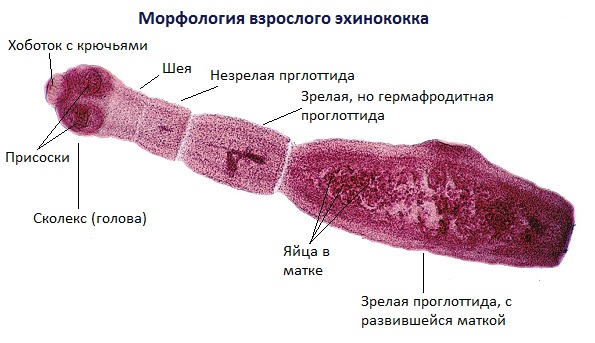
Человека поражает личиночная стадия развития Echinococcus granulosus, она долгое время растёт, живёт и развивается в организме, образуя овальной или округлой формы кисты, заполненные жидким содержимым.
Причины возникновения
Основным резервуаром инфекции являются домашние собаки. Человек заражает при контакте с животными (т.к. яйца выделяются в окружающую среду с их фекалиями, прикрепляясь к поверхности шерсти, иногда эхинококк может самостоятельно выползать из анального отверстия собаки), при употреблении в пищу продуктов питания и воды, загрязнённых яйцами паразита.
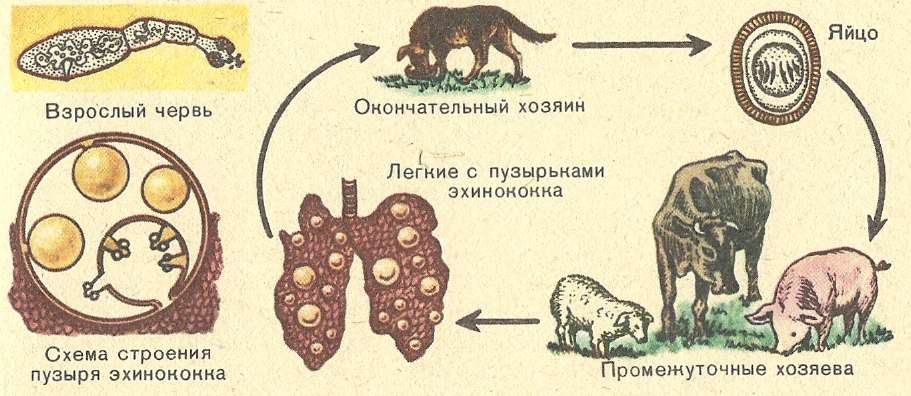
Стадии морфогенеза
Для эхинококка характерно прохождение определенных стадий в своём развитии (морфогенез):
- 1.зрелая особь;
- 2.яйцо;
- 3.личинка (онкосфера);
- 4.финна;
- 5.эхинококковый пузырь (киста).
Рассмотрим эти стадии подробнее.
Зрелая особь или её часть – сегмент, членик, обитающая в организме окончательного хозяина (домашние животные), вместе с испражнениями выходит наружу, при этом членик, содержащий яйца паразита, лопается.
Они оказываются в окружающий среде (могут долго находиться в латентном периоде, сохраняя способность к инвазии и заражению) и прикрепляются к различным поверхностям: к шерсти животным, траве, предметам, попадают в воду и диссеминируют в ней.
Эти яйца алиментарным путем или фекально-оральным попадают в организм человека, наступает инвазионная стадия развития – личинка (онкосфера). Стадия морфогенеза заключается в том, что будущая онкосфера теряет свою оболочку, адгезирует к стенке сосуда, перфорирует её, проникая в ток крови и разносится большим кругом кровообращения по организму.
Личинка может оказаться не только в органах, но и в костной ткани, мышечной, жировой. Прикрепившийся к субстрату паразит вступает в новую период развития, трансформируясь в финну, которая представляет собой большой пузырь, заполненный жидкостью и содержащий сколексы.
Эхинококковый пузырь медленно растёт, получая необходимые для жизнедеятельности вещества и может достигать огромных размеров, оказывая давления на прилежащие органы и ткани, а при прорыве стенки кисты (в результате резкого толчка, гнойного расплавления при обострении) всё его содержимое (сколексы) изливается наружу, вызывая не только новые очаги поражения, но и токсический шок.
Человек является биологическим тупиков в цикле развития эхинококка, а окончательным хозяином, то есть организмом, где формируется зрелая половозрелая особь, являются домашние и дикие животные.
Симптомы
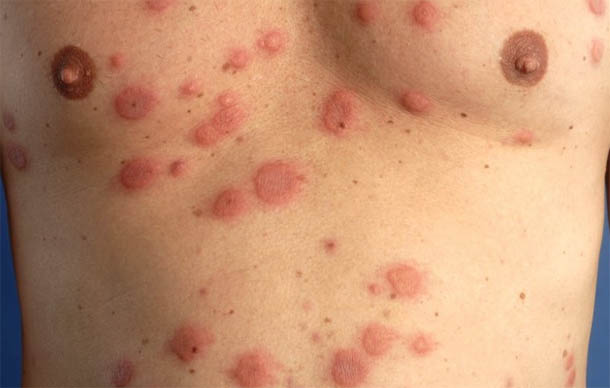
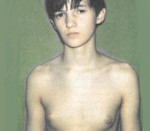
Длительное время эхинококк может не вызывать никаких симптомов у человека, если существующие кисты медленно увеличиваются в размерах, без нагноений.
Заболевание чаще диагностируется у пациентов среднего возраста, случайно, при обследовании по поводу сопутствующих заболеваний или кожной сыпи (см фото выше).
Стадийность заболевания
В клинической практике выделяют несколько условных стадий эхинококкоза:
- Латентная — развивается с момента инвазии в организм до первых появления неспецифических субъективных симптомов (плохое самочувствие, быстрая утомляемость);
- Слабовыраженные изменения— более выраженные субъективные расстройства;
- Стадия прогрессии— резко выраженные объективные признаки (сыпь, гепатомегалия);
- Стадия осложнений — нагноение и прорыв кисты с дальнейшей генерализацией процесса (возникновение вторичных очагов) и развитием интоксикации, вплоть до токсического шока.
Формы эхинококкоза, особенности и возможные осложнения
В зависимости от локализации паразита выделяют:
Эхинококкоз глазного яблока – локализуется внутри глазницы, за глазом, при увеличении кисты происходит экзофтальм (пучеглазие из-за механического давления), со временем острота зрения снижается.
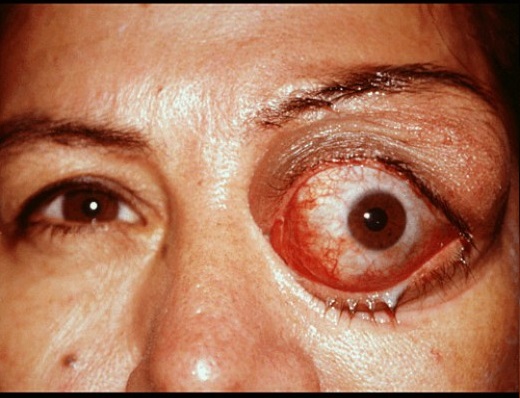
Постоянное давление кисты на глазной нерв приводит к его атрофии, возможна полная потеря зрения. Диагностика – исследование глазного дна. При обнаружении цистообразования проводится оперативное вмешательство.
Эхинококкоз головного мозга – очаг поражения может находится как в ткани мозга, так и в мозговых оболочках, заносится гематогенно. Вокруг соединительнотканной капсулы кисты нейроны подвергаются дистрофии и некрозу, наблюдаются очаги геморрагий.
Больные жалуются на постоянный сильные головные боли с рвотой и резким снижением зрения, с прогрессирующими эпилептическими припадками и неуклонным увеличением внутричерепного давления. В финальных стадиях разрушения структур мозга – бред, галлюцинации, слабоумие, деменция, судороги.
При пункции цереброспинальной жидкости обращает внимание цвет (серый) и мутность с элементами осадка, при микроскопии обнаруживаются обломки стенки кисты, конгломераты сколексов. Осложнение – эпилептический статус, кровоизлияния. Причина смерти при данном виде нозологии – внутричерепная гипертензия, инсульт.
Эхинококкоз желчевыводящих путей – первично поражение желчного пузыря, в дальнейшем развивает холангит (воспаление протоков). Больные жалуются на тошноту, рвоту, повторяющиеся приступы печеночной колики, не купирующиеся спазмолитическими препаратами, ахоличный стул, желтушность кожных покровов (в результате обтурационной желтухи).
Характерно появление мелкой папулёзной сыпи на коже, новые элементы которой доставляют пациенту неприятные субъективные ощущения – слабость, головокружение, озноб, зуд.
Эхинококкоз костей – киста располагается в костномозговой полости, приводя к пазушной резорбции, септическому остеолизису при нагноении, что становится причиной эрозий костей и их патологических переломов.
Эхинококкоз лёгкого – в одной или нескольких долях органа образуется кисты. Это вторая по частоте встречаемости – 20-30% – локализация паразита.
По мере роста, киста оказывает давление на окружающие ткани, появляется боль в загрудинной области, сухой (затем экссудативный, иногда с примесью крови) кашель, вокруг паразита формируется перифокальное воспаление, при обильной экссудации присоединяется плеврит (если воспаление не диагностируется и не лечится, возникает спаечная форма с тенденцией к развитию пневмофиброза).
Если эхинококковый пузырь достигает больших размеров, то грудная клетка может изменять свою форму, происходит выбухание межреберных промежутков.
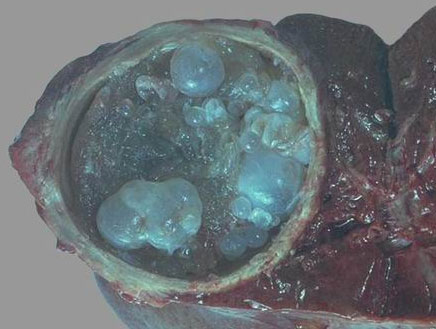
Эхинококкоз печени – составляет до 50-70% всех зарегистрированных случаев. Кисты располагаются в одной доле печени, чаще в правой, однако зафиксированы казуистические случаи билатерального поражения.
На ранних стадиях болезни отмечается боль в эпигастрии, чувство тяжести в правом подреберье.
При нагноении кисты важно провести дифференциальную диагностику эхинококкоза от абсцесса печени.
Эхинококкоз почки – чаще поражается левая почка. Различают несколько типов кист:
- Закрытая – стенка кисты интактна. Какие-либо изменения в моче не обнаруживаются, в редких случаях – незначительная гематурия (кровь в моче), протеинурия (белок в моче), в результате токсического действия паразита на почку.
- Псевдозакрытая – проникает в почечную чашечку, постоянно омывается мочой, что ведёт к изменению её качественного состава – гематурия, пиурия (лейкоциты в порции мочи).
- Открытая – часть стенки при разрушении может выявляться в суточном диурезе (эхинококкурия), так как киста сообщается с чашечно-лоханочной системой. Лейкоцитурия отмечается в 60%, гематурия — в 20% случаев; моча желто-зеленоватого цвета, мутная, с осадком в виде хлопьев и с обрывками некротизированных тканей.
Почка со временем деформируется (в зоне расположения кисты корковый слой выбухает), поверхность становится мелкозернистой, почечные чашечки и лоханки расширяются, паренхима подвергается атрофии, некрозу и дальнейшему обызвествлению. Пациентов беспокоят:
- постоянная боль в области поясницы и подреберья;
- субфебрильная температура;
- боль при мочеиспускании,
- почечная колика;
- гематурия, уменьшение суточного диуреза – вплоть до анурии.
При пальпации в нижних отделах почки выявляются круглые безболезненные эластичные образования, плотно спаянные с поверхностью, гладкие или бугристые на ощупь.
При присоединении воспаления “симптом поколачивания” может быть положительным. Лечение – резекция почки или её полное удаление.
Операции в виде выскабливания кист неэффективны. Течение благоприятное, при нагноении и диссеминировании прогноз ухудшается.
Эхинококкоз селезёнки – кисты в этой области могут быть одиночными или множественными, при последнем варианте селезёнка достигает огромных размеров, пораженная часть выбухает над поверхностью, в виде плотноэластического бугристого узла, на разрезе тёмно – вишневого цвета.
Капсула утолщенная, корковый слой атрофирован, метафоричное название патологической картины – “эхинококковый мешок”.
При разрезе обнаруживаются поликистозные разрастания дочерних кист, их стенки состоят из соединительной ткани с участками гиалиноза и оссификации, по периферии возникают васкулиты, склерозы сосудов, венозное полнокровие.
Паренхима селезёнки отёчна, с элементами геморрагии, плазморрагии и некрозов. Осложнения – при разрыве пузыря возможно развитие перитонита. Лечение – иссечение части селезёнки или полная спленэктомия.
Эхинококкоз сердца – редкая форма нозологии, регистрирующаяся в 0,2 – 2% наблюдений, болеют чаще лица старше 20 лет , мужчины.
В полости сердца (преимущественно левый желудочек) и в ткани миокарда паразиты проникают с места первичной инвазии вместе с током крови по большому кругу кровообращения.
Встречаются казуалистические случаи поражения детей с эхинококковым пузырём в перикарде и правом предсердии.
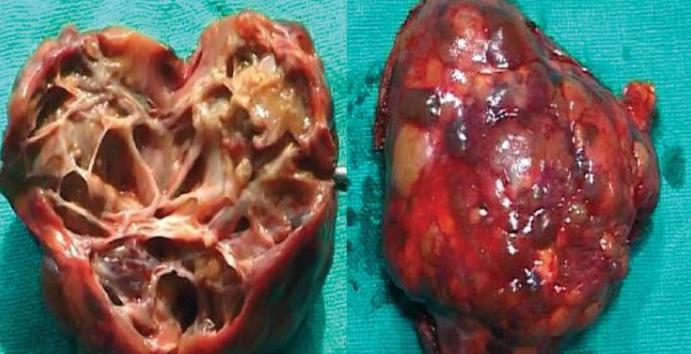
Пациенты жалуются на боли в грудной клетки, сухой кашель с кровохарканьем, выражены все признаки ишемии миокарда, поэтому важно дифференцировать эхинококкоз от хронической сердечной недостаточности с помощью инструментальных методов диагностики.
Периферический рост кист может стать причиной инфаркта миокарда из-за гнойного расплавления прилежащей стенки желудочка, к внезапной сердечной смерти из –за эмболии инородными телами (частью пузыря), гипертрофии пораженного участка с прогрессирующей декомпенсацией функциональных способностей с возможным сдавлением коронарных артерий и нарушением сердечной проводимости, обструкции выносящего тракта, тромбоэмболии, аритмии и тахикардии сердца.
В литературе встречаются случаи развития поперечной блокады сердца и полной блокады ножек пучка Хиса.
Кистообразование происходит в сравнительно быстрые сроки – от 1 до 5 лет, причём энуклеация пораженного участка не приносит значительных долгосрочных результатов, по причине множественных отсевов в другие органы и выраженной постэмболической гипертензии лёгких с синкопальным синдромом.
Эхинококкоз спинного мозга – характеризуется коротким сроком латентного периода, так как быстрый периферический рост кисты вызывает резкое сдавление протока спинного мозга с ранним развитием расстройств чувствительной и двигательной систем:
- нарушение функций конечностей (парезы, параличи);
- внутренних органов;
- изменение психики.
В связи со специфической локализацией проведение операции в виде цистизвлечения практически не представляется возможным.
Эхинококкоз позвоночника – обычно тесно связан с предыдущим вариантом, так как локализации кисты именно в телах позвонков приводит к сдавлению спинного мозга с блокадой тока ликвора и атрофией.
Длительное время симптомы не выявляются, больные жалуются на чувство тяжести в костях, клиническая картина неспецифична даже при увеличении размера паразита: опоясывающие боли в спине, в конечностях, резкие движения и кашель усиливают этот симптом.
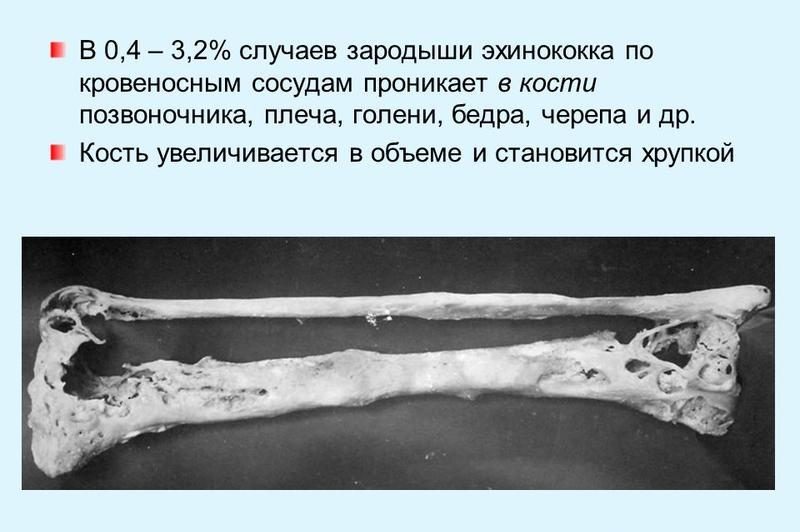
Прогрессирование заболевания ведёт к ограничению подвижности пораженных участков, деформированию костного скелета. Перкуссия костей над участком эхинококкового пузыря болезненна, при осмотре – мышцы в этой зоне валикообразно приподняты и утолщены.
Для верификации нозологии необходимо проведение рентгенодиагностики или компьютерной томографии для исключения спондилоартрита (при туберкулезе) и опухолей (остеомы, остеобластокластомы, остеосаркомы). Лечение – хирургическое.
Диагностика
Скорость нарастания симптомов при поражении человека эхинококком связана с областью образования эхинококкового пузыря, заболевание может прогрессировать до 10-20 лет , не принося пациенту никаких субъективных расстройств. Клинические проявления тоже неспецифичны:
- в крови, как и при любых гельминтозах, увеличивается уровень эозинофилов;
- повышается скорость оседания эритроцитов (СОЭ), тромбоцитов;
- появляются признаки аллергических реакций (зуд, сыпь).
При первых симптомах необходима срочная комплексная диагностика, которая заключается в инструментальных методах обследованиях – УЗИ, МРТ, КТ, пункция пораженного участка, серологический метод диагностики на выявление антител класса IgG к эхинококку.
Лечение
Консервативных способов лечения не существует! Удаление эхинококка возможно только хирургическим методом с проведением тотальной резекции пораженного органа для профилактики рецидивов.
Профилактика
Основным методом профилактики является соблюдение личной гигиены и чистоты рук.
Видеозаписи по теме
Читайте также:
